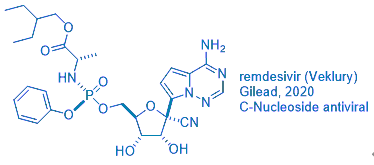
Gileadʹs antiviral drug remdesivir (Veklury) was approved by the FDA on May 1, 2020 for emergency use in coronavirus disease-2019 (COVID-19) patients after a one-day review. It took the Japanese government much longer to give the nod, seven days, before granting its regulatory approval.
But one wonders: how was remdesivir discovered?
Its genesis retraces back to the first antiviral drug, idoxuridine.
Inspired by George H. Hitchings (Nobel laureate in 1988 for Physiology and Medicines), who started to systemically investigate purine and pyrimidine analogs as potential drugs, Professor William H. Prusoff at Yale discovered idoxuridine (IdU) as the first small-molecule antiviral drug in 1959. He is now known as the godfather of modern antiviral chemotherapy.
Although IdU is too toxic to be given systemically, it is applied topically to treat eye and skin infection caused by herpes simplex virus (HSV). While its mechanism of action (MoA) is not completely elucidated, it is most likely phosphated first by kinases in both virus and normal cells to the corresponding nucleotide monophosphate (when a phosphate is attached to a nucleoside, it becomes a nucleotide), nucleotide diphosphate, and nucleoside triphosphate (NTP) sequentially. NTP is the active drug with two fates. On the one hand, when interacting with viral DNA polymerase, it terminates DNA replication and exerts antiviral activities. On the other hand, when interacting with cellular DNA polymerases, cytotoxicity, mitochondial toxicity, and antitumor activity ensued.
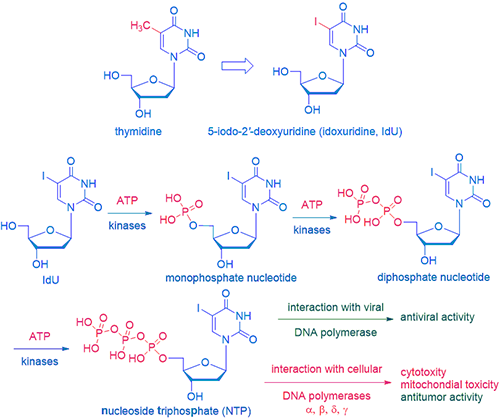
The emergence of IdU opened a floodgate of ribonucleoside antiviral drugs. It was followed by trifluorothymidine (TFT, Viroptic), ethyldeoxyuridine (EdU), bromovinyldeoxyuridine (BVDU), and more recently, telbivudine (Tyzeka), a synthetic thymidine nucleoside analog put on the market by Novartis in 2006.
Gertrude “Trudy” Elion, who shared the 1988 Nobel Prize with George Hitchings, led a team at Wellcome Research Institute that discovered acyclovir (Zovirax) for the treatment of herpes simplex virus (HSV). One of the Wellcome chemists, Howard Schaeffer, established that the intact sugar ring of compounds (such as guanosine) was not essential for binding to enzymes needed for DNA synthesis. Cutting off the diol fragment of the sugar fragment led to the discovery of acyclovir, which became one of the most successful antiviral drugs at the time. The creativity of scientists is boundless!
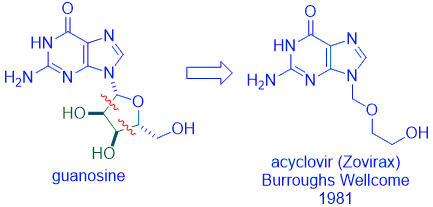
Following the discovery of acyclovir, its “me-too” drugs, ganciclovir (Cytovene, 1988) and penciclovir (Denavir, 1996), followed.
Nucleoside antiviral drugs saved the day when AIDS was a virtual death sentence in the early 1980s because there was no effective drug at all. In 1984, National Cancer Institute (NCI) began a screening program of promising drug candidates solicited from major pharmaceutical companies. Only one compound showed ability to block HIVʹs reverse transcriptase activities. It was azidothymidine (AZT, Zidovudine), submitted by Burroughs Wellcome, although it was initially synthesized by Jerome Horowitz at Wayne State University. As shown below, the only structural difference between thymidine and AZT is the hydroxyl group is replaced by an azide group. In terms of MoA, after phoshorylation by kinases, the AZT-triphosphate (TP) “pretends” to be thymidine-TP and is incorporated by the reverse transcriptase into viral DNA. When present at the end of a growing chain of DNA, it prevents additional DNA synthesis incorporation. This is why nucleoside antivirals, such as AZT, are known as chain terminators.
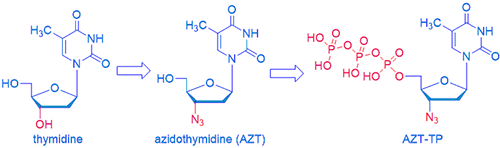
AZT is also reasonably selective. AZT-TP inhibits the synthesis of DNA by reverse transcriptase about 100-times better than it inhibits the synthesis of DNA by the host-cell DNA polymerase in the cell nucleus. More impressively, AZT inhibits HIV replication at concentrations about 1,000-fold less than it takes to inhibit the replication of the host-cell lymphocytes. In other words, AZT kills the virus faster than normal healthy cells thus providing a therapeutic window. As Hippocratic Oath states: primum non nocere: first, do no harm!
Americaʹs hero Dr. Anthony Fauci compared remdesivirʹs efficacy against COVID-19, though not overwhelming, to the discovery of AZT for HIV for a good reason. After AZT, many “me-too” and “me-better” nucleoside reverse transcriptase inhibitors (NRTIs) emerged on the market. They include d4t (Zerit), ddI (Videx), lamivudine (3TC, Epivir) discovered by Dennis Liotta at Emory, and abacavir (Ziagen) discovered by GSK and Bob Vince at Minnesota. Meanwhile, many non-nucleoside reverse transcriptase inhibitors (NNRTIs) were also discovered: nevirapine (Viramune), efavirenz (Sustiva), delavirdine (Rescriptor), etravirine (Intelence), and rilpivirine (Edurant). Concurrently, many HIV protease inhibitors, such as saquinavir (Invirase), indinavir (Crixivan), ritonavir (Norvir), tipranavir (Aptivus), and darunavir (Prezista), have been discovered as well. In 1996, highly active antiretroviral therapy (HAART), also known as cocktail HIV drugs, transformed AIDS from a death sentence to a chronic disease that can be managed with drugs (close to a cure), a scenario we hope to achieve soon for COVID.
Closer to home, HCV drugs built the foundation for the discovery of remdesivir.
Hepatitis C virus (HCV) drugs represent a major triumph of modern medicine. Different from hepatitis A virus (HAV) and hepatitis B virus (HBV), some patients affected by HCV have no symptoms at all, which explains why it was not until 1989 when HCV was cloned…even though about 200 million people worldwide are infected.
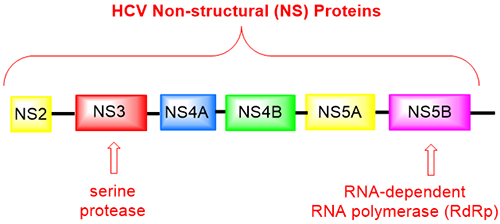
Unlike herpes simplex virus (HSV) and HIV that are DNA viruses, HCV is a single-strand RNA virus. Proteins encoded the HCV genome include several non-structure (NS) proteins. One of them is the NS5B protein, a RNA-dependent RNA polymerase (RdRp, incidentally remdesivir inhibits COVIDʹs RdRp as well), which is the weakest link of the virus genome, most vulnerable point for viral replication within the host. HCV NS5B, as other non-structural proteins such as NS3/4A and NS5A, are responsible for the replication of the viral genome that represent important and tractable targets for drug design and development.
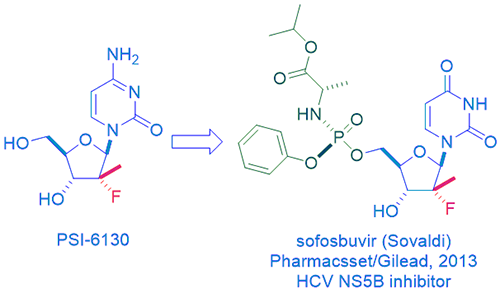
One of the shining stars of HCV NS5B inhibitors was sofosbuvir (Sovaldi), discovered by Pharmasset1 and sold by Gilead. It captured every American’s attention when it was sold at one thousand dollars a pill! It was justified since it offered a cure when there was none and the cost was still significantly less than any other alternatives at the time.
Sofosbuvir is a prodrug of a unique nucleoside PSI-6130, which is fascinating on its own right because it has a rare fluorine-containing tertiary carbon at the 2ʹ-position of its ribose ring. This is a testimony of how important that the fluorine atom has become in modern drug discovery. While no fluorine-containing drugs existed before fludrocortisone was approved in 1955, nowadays, more than 20% of all drugs contain one or more fluorine atoms. For the record, as of today, the trophy for the drug with the most fluorine atoms (seven) goes to aprepitant (Emend), a drug marketed by Merck since 2003 to prevent nausea and vomiting brought upon by cancer chemotherapy. It is a substance P antagonist and a neurokinin 1 (NK1) inhibitor.
The major aspect that made sofosbuvir significantly superior than its parent drug PSI-6130 is that the former is a prodrug of the latter. Perusal of any medicinal chemistry book, you will find that the prodrug strategy can turn a terrible drug into a decent one. The litany of prodrugsʹ benefits includes (a) Overcoming formulation and administration problems; (b) Overcoming absorption barriers; (c) Overcoming distribution problems; (d) Overcoming metabolism and excretion problems; and (e) Overcoming toxicity problems. As a consequence, prodrugs currently constitute 5% of known drugs and a larger percentage of new drugs.
Sofosbuvir is not just any prodrug, but it is the state-of-the-art phosphoramidate prodrug. Those Prodrugs of nucleoTides are known as ProTides. The technology was developed in 1990 by Professor Chris McGuigan at Cardiff University in Wales. It is probably the most successful prodrug strategy applied in the antiviral field.
In order to convert PSI-6130 to the active PSI-6130-TP, the phosphorylation has to take place first to make the 5ʹ-monophosphate of PSI-6130. However, as it so often happens, the virus either does not induce a specific kinase or has developed resistance to the compound through mutations in this enzyme while human cell fails to secure phosphorylation. To overcome this issue, it is better to install a phosphate onto the nucleoside (which is now a nucleotide since it has a phosphate). Regrettably, phosphates are negatively charged, and those nucleotides all have poor cell penetration at physiological pH. Among many tactics to overcome the polarity issue, the most successful of all is the ProTide where the nucleotide is masked with an amino acid ester pro-moiety linked via a P–N bond. The frequently used amino acid, such is the case for sofosbuvir, is L-alanine. Such a ProTide can enter the cell via facilitated passive diffusion through the cell membrane. Once inside the cell, the monophophate nucleoside is released and does what it supposed to do: serving as a viral RNA-replication terminator. Sofosbuvir is such a successful drug that it can be taken orally.
Many combined factors made sofosbuvir a successful efficacious and orally bioavailable drug. All stars are aligned for sofosbuvir. This was why Gilead spent $11 billion to buy Pharmasset. Even though Wall Street cried that it was overpriced, Gilead still made out ahead because Sovaldi was scientifically a triumph and financially a windfall.
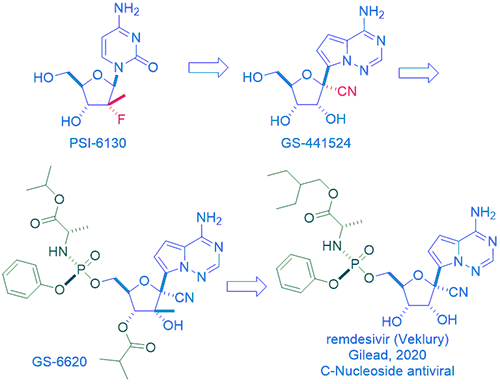
Meanwhile, Gilead came up with their own HCV NS5B drugs, one of them was nucleoside GS-44154. Gileadʹs GS-44154 has two points of differentiation in comparison to Pharmasset’s PSI-6130. One is that the former has a tertiary carbon with the key nitrile group at the 1ʹ-position on the ribose ring. More significantly, in place of the latter’s natural N-nucleoside base, the former has an unnatural C-nucleoside base, also at the 1ʹ-position on the ribose ring. Thus, GS-44154 is a 1ʹ-cyano-substituted C-nucleoside ribose analog. In theory and in practice, C-nucleosides are more resistant to metabolism in human body. In 2012, while evaluating GS-44154 in cell-based assays against a panel of RNA viruses, it was found to display broad spectrum activity against HCV, Dengue virus-2, influenza A, SAS-CoV, etc.2 SAS-CoV stands for severe acute respiratory syndrome coronavirus, which is structurally close to current SAS-CoV-2, the causative viral pathogen of COVID-19.
Most nucleosides, including GS-44154, are poorly cell-permeable, they can have a low hit rate in cell-based antiviral screens. Prodrugs are often resorted to enhance their cell permeability. For nucleoside GS-44154, after installation of a phosphoramidate, the prodrug du jour, Gilead arrived at their own HCV NS5B inhibitor, GS-6620. It was the first C-nucleoside HCV polymerase inhibitor with demonstrated antiviral response in HCV-infected patients.3 With the overwhelming success of sofosbuvir (Sovaldi), though NIH (not invented here), there was no need for another “me-too” drug for Gilead. The home-grown PSI-6130 languished on the self of Gileadʹs compound management.
Then came along the Ebola virus (EBOV), a member of the Filoviridae family. EBOV is a single-stranded, negative sense, non-segmented RNA virus. During 2010s, more than 28,000 cases of Ebola virus disease were recorded in West Africa. To combat the pandemic, Gilead collaborated with the Center for Disease Control (CDC) and the United States Army Medical Research Institute of Infectious Diseases (USAMRIID). Together, they screened an assembly of approximately 1000 compounds, largely nucleosides and nucleotides from Gilead’s collection. They heavily focused on ribose analogs that could target RNA viruses since this would encompass many emerging viral infections ranging from respiratory pathogens belonging to the Coronaviridae family such as severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), to mosquito-borne viruses of the Filoviridae family such as Dengue and Zika. What emerged from the one thousand compounds was nucleoside GS-44154 and the corresponding phosphoramidate prodrug of its monophosphate. The prodrug was the Sp isomer GS-5734, which would go on to become remdesivir and Veklury in due course.4 As any chemist would know, even a minute chemical structural change may result in tremendous differences in physiochemical and pharmacological properties. Despite striking structural similarities, remdesivir has to be given via injection, whereas sofosbuvir is orally bioavailable.
In a 2012 study, GS-5734 (remdesivir) showed antiviral activity against SAR strain Toronto 2 without cytotoxicity toward the host cells. In 2016, therapeutic efficacy of remdesivir was demonstrated against Ebola virus in rhesus monkeys, a validated animal model.5 Since the drug worked on nonhuman primates, it was deemed safe and efficacious enough to bring up to humans for clinical trials. At the time, four drugs were in clinical trials for treating the Ebola virus disease: ZMapp by Mapp Biopharmaceuticals, Mab114 by Vaccine Research Center at NIH, REGN-EB3 by Regeneron, and remdesivir by Gilead. Remdesivir is a small-molecule drug, while the other three drugs, ZMapp, Mab114, and REGN-EB3, are all monoclonal antibody (mAb) products. Monoclonal antibodies are derived from immune system molecules that bind to a specific substance, such as an invading virus. Pretty soon, ZMapp and remdesivir were dropped from the remainder of the trials because these two drugs were much less effective at preventing death than Mab114 and REGN-EB3. Eventually, it seemed that the Ebola virus pandemic was under control after ravaging West Africa for many years.
Then came along COVID-19 in Wuhan, China at the end of 2019. In terms of damages, COVID-19 takes the crown of all coronaviruses. Like Ebola virus, COVID-19 is a RNA virus. It did not take long for Gilead scientists to propose that remdesivir could be a potential treatment of COVID-19 very early on. First of all, remdesivir showed inhibitory effects on pathogenic animal (rhesus macaques) and human coronaviruses, including SARS-CoV-2, the causative viral pathogen of COVID-19, in vitro by inhibiting its RNA-dependent RNA polymerase (RdRp). It also inhibits SARS and MERS coronaviruses.
Ironically, remdesivir sounds like “people’s hope” in Chinese phonetically. During the dark days of initial pandemic, remdesivir was people’s only hope.
Although the outcome of clinical trials for remdesivir in China was ambiguous, Fauciʹs team at National Institute of Allergy and Infectious Diseases (NIAID) found statistically significant benefits of remdesivir for treating COVID patients using the gold standard of clinical trials: randomized and double-blinded clinical trials. The rest, like they say, is the history.6
If there is a silver-lining of this COVID-19 pandemic, I hope it is the renewed realization of the importance of science and medicine. A new generation of youth would choose a career in STEM or medicine to save peopleʹs lives over other more lucrative careers. We will create real medicines to cure diseases, not opting to drink disinfectants to kill the virus.
References
- Sofia, M. J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P. G.; Ross, B. S.; Wang, P.; Zhang, H.-R.; et al. J. Med. Chem. 2010, 53, 7202–
- Cho, A.; Saunders, O. L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J. E.; Feng, J. Y.; Ray, A. S.; Kim, C. U. Med. Chem. Lett. 2012, 22, 2705–2707.
- Cho, A.; Zhang, L.; Xu, J.; Lee, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M.; et al. Med. Chem. 2014, 57, 1812–1825.
- Siegel, D.; Hui, H. C.; Doerffler, E.; Clarke, M. O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Med. Chem. 2017, 60, 1648–1661.
- Warren, T. K.; Soloveva, V.; Wells, J.; Stuthman, K. S.; Van Tongeren, S. A.; Garza, N. L.; Donnelly, G.; Shurtleff, A. C; Retterer, C. J; Gharaibeh, D.; et al. Nature 2016, 531, 381–
- Eastman, R. T.; Roth, J. S.; Brimacombe, K. R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M. D. ACS Cent. Sci. 2020, in press.


