This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

A Versatile and Sustainable Multicomponent Platform for the Synthesis of Protein Degraders: Proof-of-Concept Application to BRD4-Degrading PROTACs
Authors: Irene Preet Bhela, Alice Ranza, Federica Carolina Balestrero, Marta SerafiniSilvio Aprile, Rita Maria Concetta Di Martino, Fabrizio Condorelli, Tracey Pirali
Abstract
The use of small molecules to induce targeted protein degradation is increasingly growing in the drug discovery landscape, and protein degraders have progressed rapidly through the pipelines. Despite the advances made so far, their synthesis still represents a significant burden and new approaches are highly demanded. Herein we report an unprecedented platform that leverages the modular nature of both multicomponent reactions and degraders to enable the preparation of highly decorated PROTACs. As a proof of principle, our platform has been applied to the preparation of potential BRD4-degrading PROTACs, resulting in the discovery of a set of degraders endowed with high degradation efficiency. Compared to the existing methods, our approach offers a versatile and cost-effective means to access libraries of protein degraders and increase the chance of identifying successful candidates.
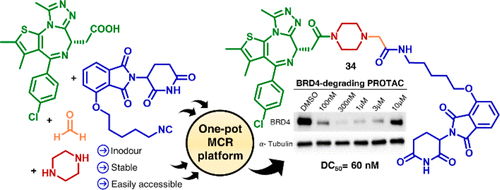
Overview
From I. P. Bhela, T. Pirali et al.. After an informative review on the state of the art for synthesis of PROTACs, this report offers up a 1 step approach to finished PROTACs from multicomponent Ugi or Passerini reactions. The POI binder just needs to have a CO2H ready to react and the E3 ligase binder needs a preformed linker terminated by an -NC group. This has the potential to make entry into this field easier for newcomers. Here is an example:
Enhancing the Safety and Efficacy of PSMA-Based Small-Molecule Drug Conjugates by Linker Stabilization and Conjugation to Transthyretin Binding Ligand
Authors: Toufiq Ul Amin, Rasha Emara, Arindom Pal, Hala Aldawod, Guanming Jiang, Dengpan Liang, Md Tariqul Haque Tuhin, Abdulmalek Balgoname, Arjun D. Patel, Mamoun M. Alhamadsheh
Abstract
This work describes the enhancement of a novel antitumor therapeutic platform that combines advantages from small-molecule drug conjugates (SMDCs) and antibody drug conjugates (ADCs). Valine–citrulline (VCit) dipeptide linkers are commonly used cathepsin B cleavable linkers for ADCs. However, the instability of these linkers in mouse serum makes translating efficacy data from mouse to human more challenging. Replacing the VCit linker with glutamic acid–valine–citrulline (EVCit) has been reported to enhance the stability of ADCs in mouse serum. However, the effect of EVCit linker on the stability of SMDCs has not been reported. Here, we report that incorporating the EVCit linker in prostate-specific membrane antigen-targeting SMDCs, equipped with the transthyretin ligand AG10, resulted in conjugates with lower toxicity, an extended half-life, and superior therapeutic efficacy to docetaxel in a xenograft mouse model of prostate cancer. This should make SMDCs’ preclinical toxicity and efficacy data from mice more reliable for predicting human results.
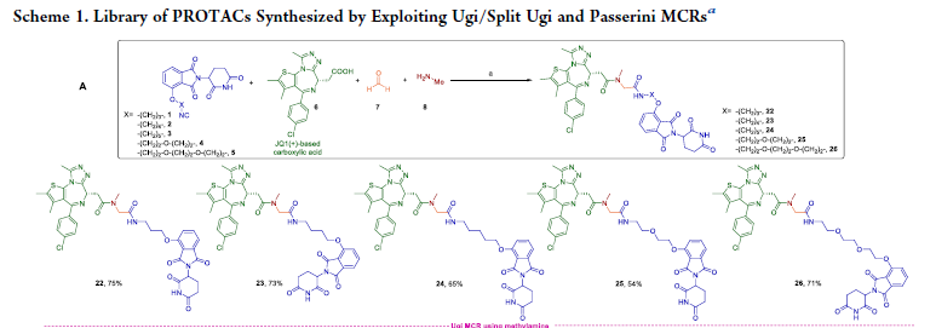
Overview
From T. U. Amin, M. M. Alhamadsheh et al. This is an advance in small-molecule-drug conjugates (SMDCs) which when compared to ADCs should have better cancer tissue penetrating properties. The functionality glutamic acid-valine-citrulline (EVCit) was introduced to provide stability to mouse extracellular serum carboxylesterase 1c (Ces1c) so that mouse xenograft cancer assays can be studied in preclinical research.

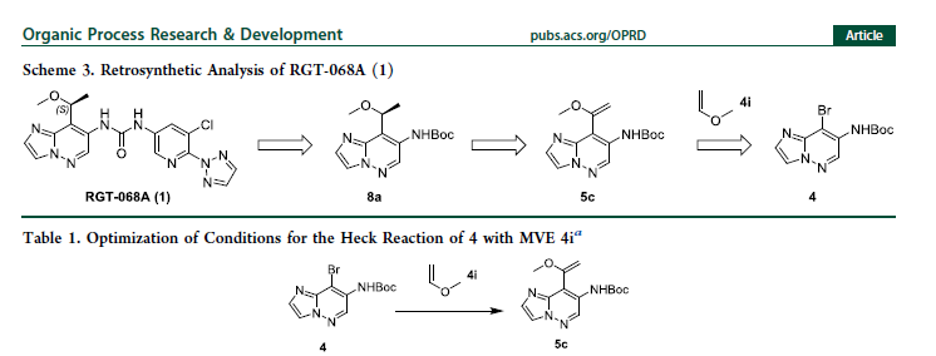
Direct Construction of Chiral Ether via Highly Efficient Heck Reaction and N-Directed Asymmetric Hydrogenation for Large-Scale Synthesis of MALT1 Inhibitor RGT-068A
Authors: Song Feng, Haishen Zhang, Zhiyong Tang, Xingao Peng, Min Yang, Xudong Wei, Wenge Zhong
Abstract
Chemical process development efforts leading to the large-scale production of RGT-068A are discussed. Process optimization resulted in (1) successful replacement of the Stille coupling reaction involving toxic stannane reagent via highly efficient Heck reaction of methyl vinyl ether, (2) construction of the chiral ether unit through N-directed Ru-catalyzed asymmetric hydrogenation avoiding super-critical fluid chromatography (SFC) chiral separation, and (3) a streamlined process with a significantly improved overall yield of about 25%, 5-fold higher than the original discovery synthetic route, which enabled the delivery of high-quality material for IND-enabling studies.
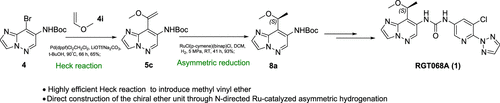
Overview
From S. Feng et al. This work presents a clever approach to a chiral angular methyl geminal to a methoxy-group. Sometimes an angular methyl like this is introduced to help with the stability of the methoxybenzyl group. This seems like a valuable small group for medicinal chemistry.

N-Trifluoropropylation of Azoles through N-Vinylation and Sequential Hydrogenation
Authors: Shu-Jie Chen, Jia-Hui Li, Zhi-Qing He, Guo-Shu Chen, Yin-Yin Zhuang, Chang-Ping Chen, Yun-Lin Liu
Installing a fluoroalkyl group onto the nitrogen atom of azoles represents a potential strategy for lead optimization in medicinal chemistry. Herein, we describe a method for the N-trifluoropropylation of azoles. This process is accomplished using a combination of regioselective N-vinylation and sequential hydrogenation. The two-step sequence is applicable to a diverse set of azoles and tolerates a wide range of functionalities. In addition, we showcase its practicability and utility through the gram-scale synthesis and the late-stage modification of a complex molecule.
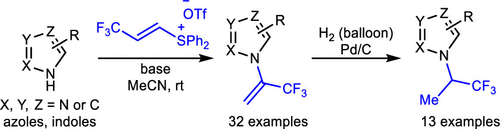
Overview
From S.-J. Chem, Y.-L. Liu et al. Similar to the other paper this one shows how to introduce CH(CF3)CH3 onto an azole NH position. This is another small group that may resist metabolism. The list of azoles includes tetrazoles, pyrazoles, imidazoles and others. With some but not all there is an issue of regioselectivity.
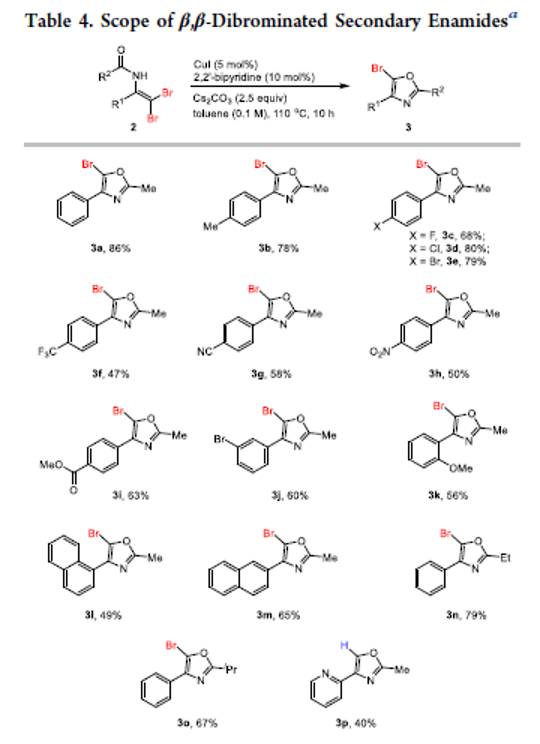
Development of β,β-Dibrominated Secondary Enamides and Their Application to the Synthesis of 5-Br Oxazoles
Authors: Shu-Jie Chen, Jia-Hui Li, Zhi-Qing He, Guo-Shu Chen, Yin-Yin Zhuang, Chang-Ping Chen, Yun-Lin Liu
Herein, we report a base promoted dibromination of enamides using CBr4 as a bromine source to provide expedient access to β,β-dibrominated secondary enamides. Preliminary synthetic application study shows that these novel products can be readily transformed to 5-Br oxazoles via Cu(I) catalyzed intramolecular cyclization in good yields.
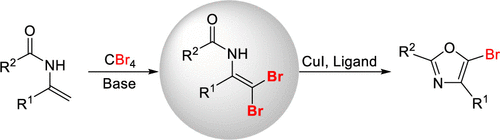
Overview
From C. Wang, F. Li et al. From enamides to 5-bromo-oxazoles. This could prove useful.
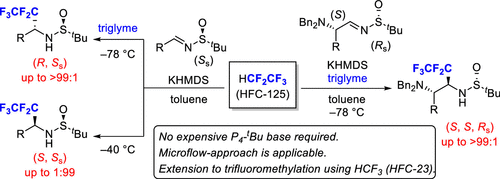
KHMDS/Triglyme Cryptate as an Alternative to Phosphazene Base in Stereodivergent Pentafluoroethylation of N-Sulfinylimines Using HFC-125
Authors: Yuji Sumii, Hiroto Iwasaki, Yamato Fujihira, Elsayed M. Mahmoud, Hiroaki Adachi, Takumi Kagawa, Dominique Cahard, Norio Shibata
Abstract
A protocol for the stereodivergent pentafluoroethylation of N-sulfinylimines using HFC-125 with KHMDS/triglyme has been developed. Both diastereomers of the pentafluoroethylated amines can be selectively synthesized based on the presence or absence of triglyme. This additive-controlled protocol allows the KHMDS/triglyme cryptate to be a straightforward and cheap alternative to previously reported base-controlled stereodivergent trifluoromethylation using potassium hexamethyldisilazide (KHMDS) versus P4–tBu.
Overview
From Y. Sumii, N. Shibata et al. A strong base system deprotonates HCF2CF3 and it adds into the chiral sulfimmine to make in the end chiral CF3CF2-CHRNH2, another useful F-containing small building block.


