Gileadʹs antiviral drug remdesivir (Veklury, 1) was approved by the FDA on May 1st for emergency use against coronavirus disease-2019 (COVID-19) after just a one-day review. Now the onerous task falls upon Gilead to make enough of the drug to supply doctors and patients around the world.
But one wonders: how is remdesivir made?
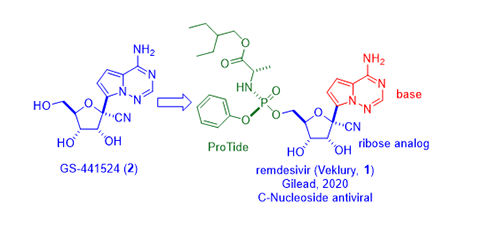
Remdesivir (1) is a ProTide, a phosphoramidate prodrug of nucleoside GS-44154 (2), which consists of two parts: the ribose core and the unnatural base. Two generations of synthesis of remdesivir (1) have been reported by Gilead.
a. The First Generation Synthesis
Gilead published two papers in 2012, one on medicinal chemistry,1 and the other on a practical synthesis of GS-44154 (2) and its analogs.2
As shown in the scheme below, pyrrolotriazine-bromide 4 was prepared according to a Bayer patent (vide infra).3 The bis-silylchloride 5 was a “transient” protective group. It capped the amine group on pyrrolotriazine-bromide 4 first, which would allow the subsequent halogen-metal exchange between the bromide 4 and n-BuLi. To the lithiated intermediate was added commercially available benzyl protected ribonolactone 3, giving rise to adduct lactol 6 as a mixture of diastereomers at the 1ʹ-position. The bis-silyl protective group was removed during acidic workup. Subsequently, in the presence of a Lewis acid (BF3•OEt2), lactol 6 was treated with TMS-CN to provide nitrile 7 as a mixture of 85:15 ratio favoring the b-anomer. BCl3-promoted debenzylation provided the desired nucleoside as GS-44154 (2) after chromatographic separation of the two diastereomers.1,2
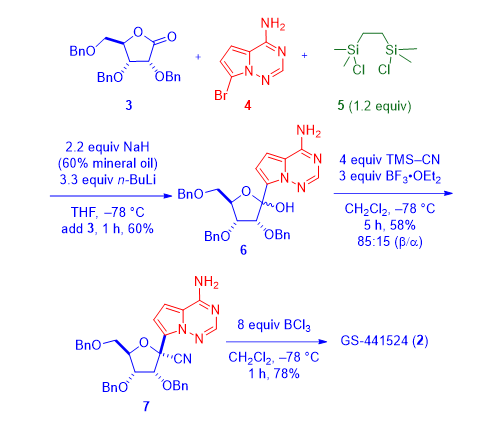
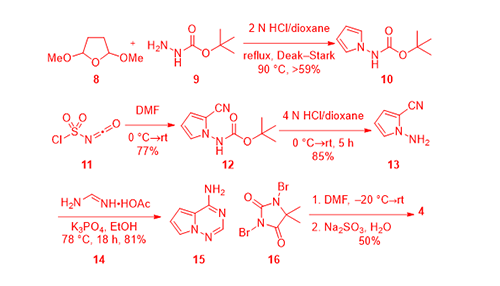
As eluded earlier, pyrrolotriazine 4 was prepared according to a Bayer patent that covered insulin-like growth factor 1 receptor (IGF-1R) kinase inhibitors for treating cancer.3 To that end, condensation between 2,5-dimethoxytetrahydrofuran (8) and tert-butylcarbazate (9) in the presence of 2 N HCl gave pyrrole 10. Treatment of pyrrole 10 with chlorosulfonyl isocyanate (11) installed the nitrile group onto the 2-position of the pyrrole ring to produce 12. Acidic removal of the Boc protective group on 12 afforded 13, which was then condensed with formamidine acetate (14) to assemble the key intermediate pyrrolotriazine 15. Bromination was accomplished using 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 16) to prepare bromide 4.3
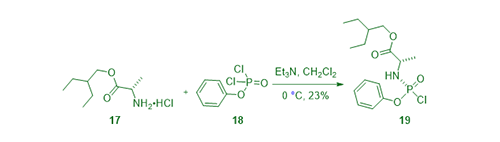
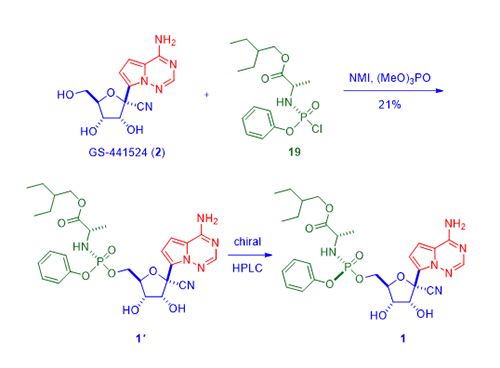
Meanwhile, the prodrug piece was prepared by treating alanine ester 17 with phenyl phosphorodichloridate (18) in the presence of Et3N to give chloridate 19, ready to be used in a further coupling reaction with nucleoside GS-44154 (2).
Finally, coupling between GS-44154 (2) and chloridate 19 was facilitated by N-methylimidazole and trimethyl phosphate to assemble phosphoramidate prodrug 1ʹ as a mixture of diastereomers. Separation using chiral HPLC then delivered the desired Sp stereoisomer remdesivir (1). Obviously, the use of chiral HPLC greatly limited the amount of the drug that can be produced and 50% of the compounds had to discarded, not only a waste of resources, but also an environmental hazard. While this synthesis is sufficient for medicinal chemists focusing on drug discovery, it is far from satisfactory when a large amount of the drug is needed, like right now. A better and cheaper manufacturing route with less waste is required to achieve that purpose. This is the job of Process Chemists.
The job of medicinal chemists is to explore structure–activity relationship (SAR) by making many compounds to probe where changes can provide a better pharmacological, physiochemical, and toxicological outcome. Therefore, a modular approach would offer more opportunities to make more compounds. A linear synthetic route is also acceptable for drug discovery as not much material is required to access its enzymatic and cellular assays, as well as determine its drug metabolism and pharmacokinetic (DMPK) profile. On the other hand, once a drug candidate reaches the hands of Process Chemists, its structure is pretty much “set”, at least the chemotype is “fixed”. Therefore, a more convergent route is preferred. Chromatography should be avoided or minimized. Green chemistry is favored since we want to generate as little waste as possible.
b. The Second Generation Synthesis
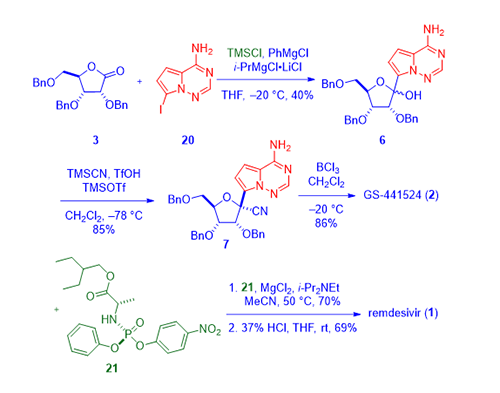
In Gileadʹs second generation synthesis of remdesivir (1),4 aminopyrrolotriazine iodide 20 was employed in place of bromide 4. After aminopyrrolotriazine iodide 20 was protected with two trimethylsilyl groups, the halogen–metal exchange was accomplished using the “turbo Grignard reagent” (isopropylmagnesium chloride–lithium chloride complex) invented by Knochel in 2004.5 Adding the cold solution of the resulting organometallic intermediate to a cold solution of lactone 3 assembled lactol 6 after quenching the reaction with a weakly acidic aqueous solution with a concurrent release of the bis-TMS protective group upon workup. In the presence of a Lewis acid (TMS–OTf), lactol 6 was treated with TMS–CN to provide nitrile as a mixture favoring the b-anomer 7. BCl3-promoted debenzylation provided the desired nucleoside GS-44154 (2). In this case, GS-44154 (2) was treated with optically pure Sp phosphoramidate 21 to deliver remdesivir (1) without resorting chiral HPLC separation.
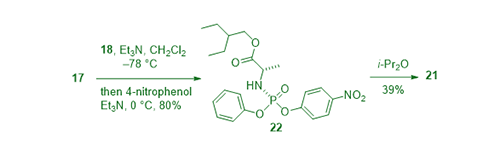
The optically pure Sp stereoisomer of phosphoramidate 21 was prepared also from alanine ester 17 and phenyl phosphorodichloridate (18) in the presence of Et3N to give the chloridate. Treating the resulting chloridate intermediate with 4-nitrophenol gave rise to phosphoramidate 22 as a mixture of two diastereomers, which was purified via recrystallization using diisopropyl ether to afford the Sp stereoisomer 21 in 39% yield, ready to use for coupling with GS-44154 (2) to make the optically pure remdesivir (1).4,6
What we have shown are merely published synthetic routes to make remdesivir (1). With highly skilled process chemists at Gilead and elsewhere, many shorter, cheaper, and greener process routes will surely emerge in the future.
References
- Cho, A.; Saunders, O. L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J. E.; Feng, J. Y.; Ray, A. S.; Kim, C. U. Med. Chem. Lett. 2012, 22, 2705–2707.
- Metobo, S. E.; Xu, J.; Saunders, O. L.; Butler, T.; Aktoudianakis, E.; Cho, A.; Kim, C. U. Tetrahedron Lett. 2012, 53, 484–
- OʹConnor, S. J.; Dumas, J.; Lee, W.; Dixon, J.; Cantin, D.; Gunn, D.; Burke, J.; Phillips, B.; Lowe, D.; Shelekhin, T.; et al. WO200756170 (2007).
- Siegel, D.; Hui, H. C.; Doerffler, E.; Clarke, M. O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Med. Chem. 2017, 60, 1648–1661.
- Krasovskiy, A.; Knochel, P. Chem. Int. Ed. 2004, 43, 3333–3336. (b) Bao, R. L.-Y.; Zhao, R.; Shi, L. Chem. Commun. 2015, 51, 6884–6900.
- Clarke, M.; OʹNeil, H.; Feng, J. Y.; Jordan, R.; Mackman, R. L.; Ray, A. S.; Siegel, D. US Pat. 10251904B2 (2017).


