Gileadʹs antiviral drug remdesivir has been catapulted to the limelight as the only FDA-approved treatment of coronavirus Covid-19 for emergency use thus far. On April 29, 2020, preliminary clinical trials indicated that the drug has shown some efficacy. While mortality rate for patients taking remdesivir is 8%, the control groupʹs mortality rate is 11%. More meaningfully, it took 11 days for patients taking remdesivir to recover while it took 15 days for patients on placebo. Finally, remdesivir is the first ray of sunshine in the dark clouds casted by the deadly invisible coronavirus. It took merely one day for the FDA to make a decision: on May 1, 2020, the FDA authorized remdesivir for emergency use in COVID-19 patients.
But one wonders: how does remdesivir work?
Remdesivir1, a bioisostere of natural N-nucleosides such as adenosine and guanosine, is a C-nucleoside antiviral drug, which works as an antimetabolite and blocks the synthesis of viral DNA.
There are four DNA bases: adenine, guanine, thymine, and cytosine. Another base, uracil, is only seen in RNAs. Remdesivirʹs1 pyrrolotriazine fragment bears a striking resemblance to adenine and guanine. Therefore, pyrrolotriazine can serve as a bioisostere of those two bases required for the synthesis of viral DNAs.
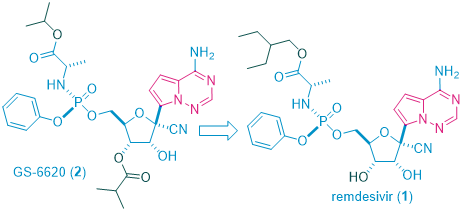
The discovery of remdesivir (GS-57341) took a long and winding route. Initially discovered as a treatment of Ebola virus (EBOV)1, it evolved from GS-66202, the first C-nucleoside HCV polymerase inhibitor with demonstrated antiviral response in HCV-infected patients.2 C-Nucleosides have the potential for improved metabolism and pharmacokinetic properties over their natural N-nucleoside counterparts due to the presence of a strong carbon–carbon glycosidic bond and a non-natural heterocyclic base.
How does remdesivir1 work as an antiviral drug? Well, it is a long story……
In terms of mechanism of action (MoA), remdesivir1 belongs to a class of nucleoside antiviral drugs as antimetabolites. By definition, antimetabolites are drugs that interfere with normal cellular function, particularly the synthesis of DNA that is required for replication. In another word, remdesivir1 “pretends” to be a nucleoside building block and participates in the viral DNA synthesis. But since it is not a bona fide DNA building block, DNA replication is terminated, and the virus is stopped from replicating.
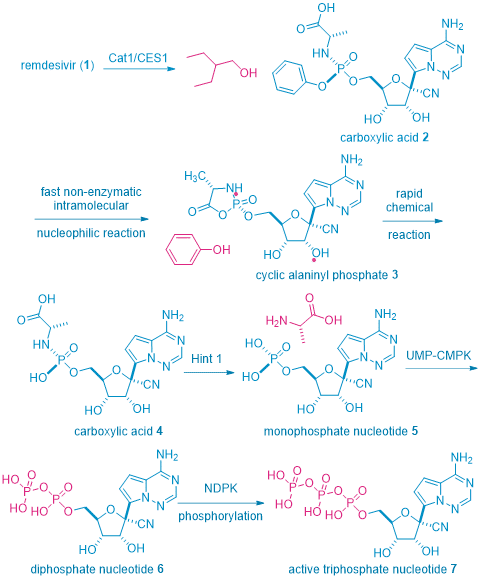
However, the reality is not that simple, remdesivir1 is a prodrug that is only active in vivo in the form of its metabolite: triphosphate nucleoside 7. But 7 has a low solubility and low bioavailability and could even be toxic. To overcome these shortcomings, the pro-drug tactic was resorted, but with a twist. To achieve optimal pharmacokinetic and pharmacodynamic outcome, several pro-drug tactics are installed onto triphosphate nucleoside 7. Therefore, remdesivir1 may be considered as a pro-drug of a pro-drug of a pro-drug of triphosphate nucleoside 7, the active antimetabolite.
As shown in the scheme beneath, once remdesivir1 is injected via IV to human body, the key enzymes initially involved in the metabolism of remdesivir1 are human cathepsin A 1 (CatA1) and arboxylesterase 1 (CES1), which are responsible for the hydrolysis of the carboxyl ester between the alaninyl moiety and 2-ethyl-2-butanol. This stereospecific reaction gives rise to the corresponding carboxylic acid 2. A non-enzymatic intramolecular nucleophilic attack then results in the formation of an alaninyl phosphate intermediate 3, which undergoes a rapid chemical reaction to hydrolyze the cyclic phosphate to a linear phosphate as carboxylic acid 4, along with a phenol. The next step is speculated to involve the histidine triad nucleotide-binding protein 1 (Hint 1) enzyme in which the alaninyl phosphate intermediate is deaminated to form a monophosphate nucleotide 5. The final two steps involve consecutive phosphorylation reactions mediated by cellular kinases, uridine monophosphate–cytidine monophosphate kinase (UMP–CMPK), and nucleoside diphosphate kinase (NDPK), producing the diphosphate nucleotide 6 and subsequently the active triphosphate nucleotide 7, respectively.3 Triphosphate nucleotide 7 is the nucleoside antimetabolite and is incorporated to the viral DNA to stop its replication.
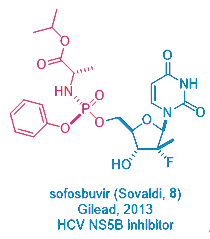
This prodrug strategy was not created from vacuum. In fact, Gileadʹs mega-blockbuster HCV drug sofosbuvir (Sovaldi, 8), licensed from Pharmasset, employs the same prodrug tactic. But Sovaldi is orally bioavailable, remdesivir1 has to be given via injection.
Now, with the FDAʹs approval, Gilead is ramping up remdesivirʹs1 production, made easier by their process chemistsʹ heroic efforts in optimizing the manufacturing route. Doctors can start prescribing remdesivir1 for emergency use in COVID-19 patients now and more and more data will accumulate on remdesivir’s1 safety and efficacy profile in a large population of patients.
Remdesivir1 is not a silver bullet, but with its proof-of-concept (PoC) in treating Covid-19 patients, we know this mechanism works. “Now this is not the end. It is not even the beginning of the end. But it is, perhaps, the end of the beginning,” to quote Churchillʹs famous words. It is expected that many “me-too,” hope fully “me-better,” drugs and innovative drugs with novel MoAs are to follow that will enable combination therapies with better safety and efficacy profiles. In addition, one day, one or more safe and efficacious vaccines will be available, fingers crossed, and we will be able to treat Covid-19 as a common flu.
References
- Siegel, D.; Hui, H. C.; Doerffler, E.; Clarke, M. O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. J. Med. Chem. 2017, 60, 1648–1661.
- Cho, A.; Zhang, L.; Xu, J.; Lee, R.; Butler, T.; Metobo, S.; Aktoudianakis, V.; Lew, W.; Ye, H.; Clarke, M.; et al. J. Med. Chem. 2014, 57, 1812–1825.
- Murakami, E.; Tolstykh, T.; Bao, H.; Niu, C.; Steuer, H. M.; Bao, D.; Chang, W.; Espiritu, C.; Bansal, S.; Lam, A. M.; Otto, M. J.; Sofia, M. J.; Furman, P. A. J. Biol. Chem. 2010, 285, 34337–34347. (b) Ma, H.; Jiang, W.-R.; Robledo, N.; Leveque, V.; Ali, S.; Lara-Jaime, T.; Masjedizadeh, M.; Smith, D. B.; Cammack, N.; Klumpp, K.; Symons, J. J. Biol. Chem. 2007, 282, 29812–29820.



