This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

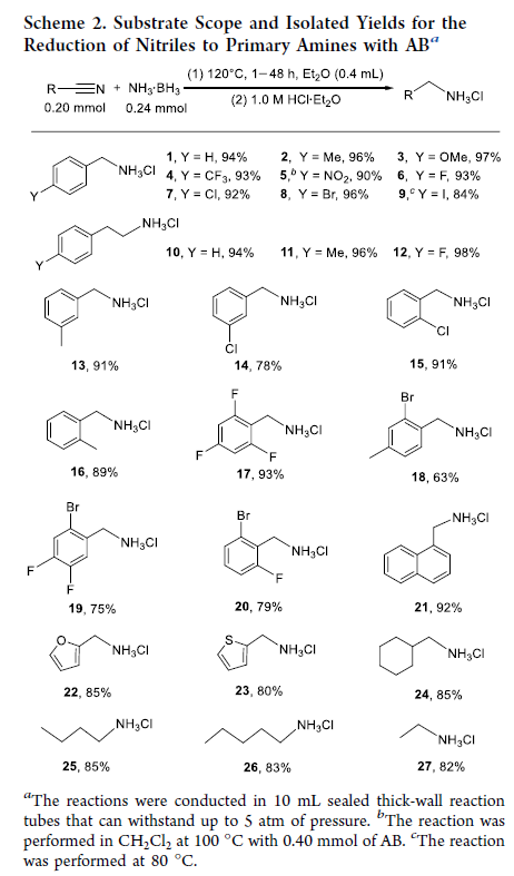
Noncatalyzed Reduction of Nitriles to Primary Amines with Ammonia Borane
Authors: Man Ding, Jiarui Chang, Jia-Xue Mao, Jie Zhang, and Xuenian Chen
The preparation of primary amines from nitriles has been a subject of continuing interest, and many different methods have been reported for this process. We report in this paper an alternative method for transforming nitriles into primary amines. In this work, a wide range of nitriles were reduced to primary amines by 1.2 equiv of ammonia borane under thermal decomposition conditions without any catalyst and the corresponding primary amines were isolated in good to excellent yields. The reactions are environmentally benign with H2 and NH3 generated as byproducts. The reactions are also tolerant of many functional groups. Nitriles are likely reduced by the in situ-generated aminodiborane, the application of which in organic synthesis has never been reported before. By using our protocol, primary amines containing multifluorinated aromatic rings, which are greatly important in pharmaceutical synthesis and have rarely been prepared via catalytic processes, were successfully prepared.
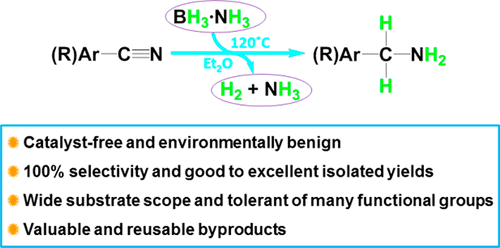
Overview
From M. Ding, J. Zhang, X. Chen et al. Use of BH3-NH3 to reduce Ar-CN or Alkyl-CN.
Examples:

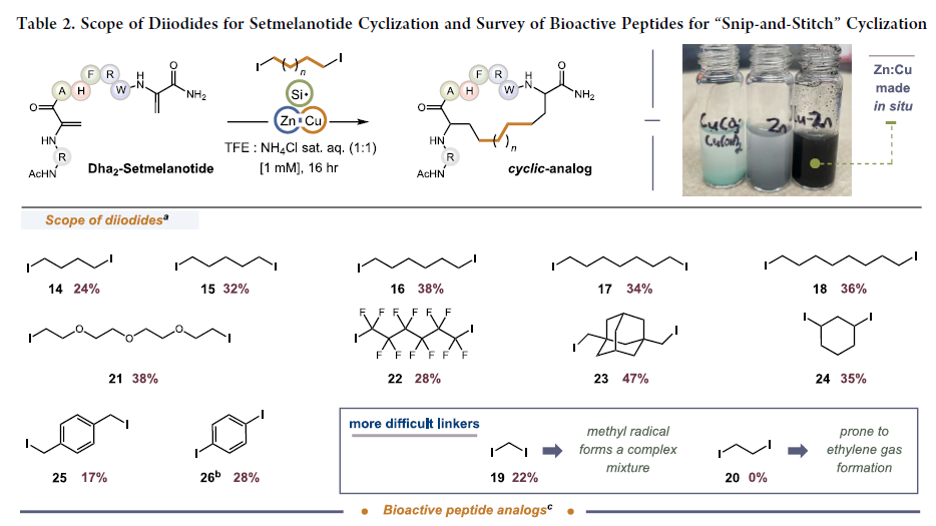
Peptide Carbocycles: From −SS– to −CC– via a Late-Stage “Snip-and-Stitch”
Authors: Samuel Gary and Steven Bloom*
Abstract
One way to improve the therapeutic potential of peptides is through cyclization. This is commonly done using a disulfide bond between two cysteine residues in the peptide. However, disulfide bonds are susceptible to reductive cleavage, and this can deactivate the peptide and endanger endogenous proteins through covalent modification. Substituting disulfide bonds with more chemically robust carbon-based linkers has proven to be an effective strategy to better develop cyclic peptides as drugs, but finding the optimal carbon replacement is synthetically laborious. We report a new late-stage platform wherein a single disulfide bond in a cyclic peptide can serve as the progenitor for any number of new carbon-rich groups, derived from organodiiodides, using a Zn:Cu couple and a hydrosilane. We show that this platform can furnish entirely new carbocyclic scaffolds with enhanced permeability and structural integrity and that the stereochemistry of the new cycles can be biased by a judicious choice in silane.
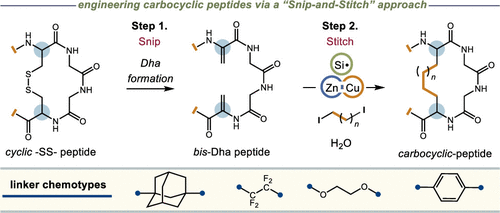
Overview
From S. Gary and S. Bloom. They call this snip and stitch and use it for macrocyclic peptide applications. However, this could have broad application as an alternative to the use of the RCM reaction for the synthesis of macrocycles. A great feature is that the inserted bridging group can be so many different things.
See the following table:


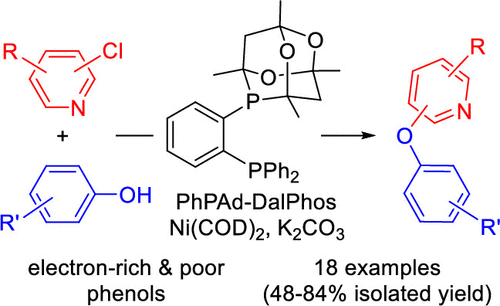
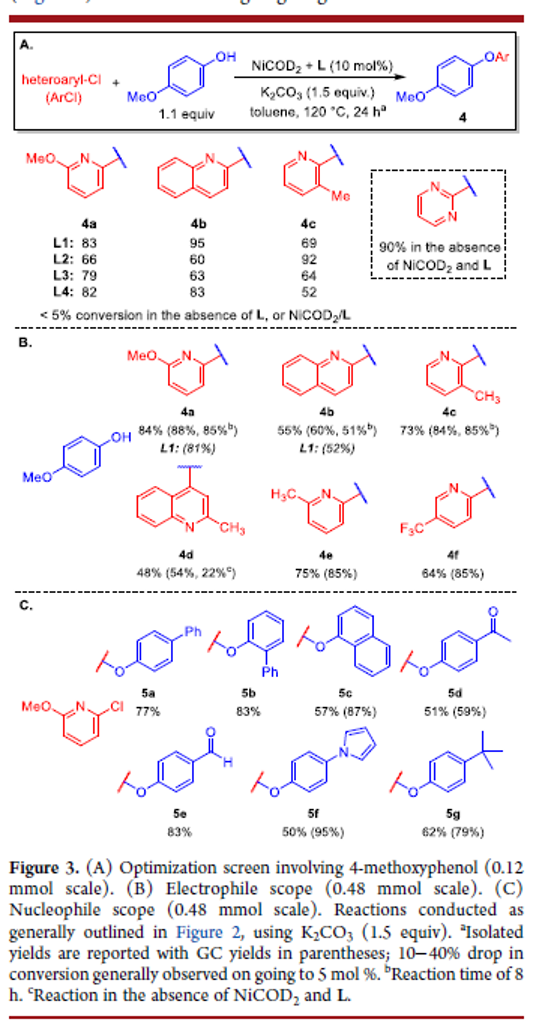
Bisphosphine/Nickel-Catalyzed C–O Cross-Coupling of Phenols with Chloropyridine and Related Electrophiles
Authors: Nicholas E. Bodé, Ryan T. McGuire, and Mark Stradiotto
Abstract
Notwithstanding recent developments in nickel-catalyzed C–O cross-coupling chemistry, such transformations of substituted phenols and (hetero)aryl chlorides with a useful reaction scope have yet to be reported. In this work, we disclose the results of catalyst screening that allowed the identification of PhPAd-DalPhos/NiCOD2 as an effective catalyst system under thermal conditions for the O-arylation of substituted phenols with chloropyridine-type electrophiles, leading to pyridyl-O-aryl frameworks that are found in active pharmaceutical ingredients.
Overview
From N. Bode, R. T. McGuire and M. Stradiotto. A Ni catalyzed phenol addition to pyridyl-halides. This would seem to be possible with simple CO3= base and polar solvent aggressively heated but the paper says no, that doesn’t work. So here you go, an alternative to the SNAr.
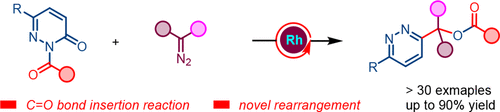
Rhodium-Catalyzed Formal C═O Bond Insertion and Sequential Acyl 1,4-N-to-O Migratory Rearrangement
Authors: Qiongya Li, Chunlan He, Jiahui Su, Ying Shao, Shengbiao Tang, and Jiangtao Sun
Abstract
We present here a rhodium-catalyzed reaction between N-acyl pyridazinones and diazoacetates, leading to pyridazine derivatives in good yield under mild reaction conditions. This whole sequence probably proceeds through a carbene insertion into a C═O bond and an unprecedented 1,4-N-to-O acyl rearrangement reaction.
Overview
From Q. Li, J. Sun et al. A clever Rh-catalyzed insertion of a carbene in between an oxo-function and a hetaryl. Of course, there needs to also be an acyl on the hetaryl-N (here as a pyridazine), but that nicely migrates onto the inserted carbene. So it is a somewhat specialized entry to 2-substituted pyridazines. Examples:
Carbene Routes to Cyclopropatetrahedrane
Authors: Murray G. Rosenberg and Udo H. Brinker
Abstract
The formation of cyclopropatetrahedrane (tetracyclo[2.1.0.01,3.02,4]pentane) via four different carbene reactions is computed using the (U)CCSD(T)(full)/cc-pVTZ//(U)ωB97X-D/cc-pVTZ + 1.3686(EZPVE) theoretical model. Intrinsic reaction coordinate plots confirm that each carbene is directly linked to cyclopropatetrahedrane via a unique cyclopropanation step. Each elementary step is assessed according to the structure and energy of its transition state.
Overview
From M. G. Rosenberg, U. H. Brinker. Cyclopropatetrahedrane! Okay, someday someone will probably make this molecule, but for now it is just the subject of calculations. Like one of those transuranium elements that everybody was after in the 30s and 40s, even if you just got a glimpse of it you could carve your name into the history books and maybe get it onto the Periodic Table. Maybe molecules like this are our transuranium elements.
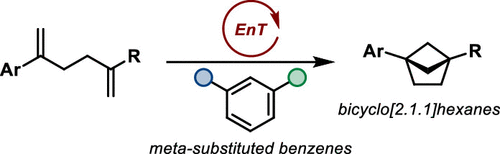
Bicyclo[2.1.1]hexanes by Visible Light-Driven Intramolecular Crossed [2 + 2] Photocycloadditions
Authors: Thomas Rigotti, Thorsten Bach
Abstract
Bicyclo[2.1.1]hexanes have become increasingly popular building blocks in medicinal chemistry as bridged scaffolds that provide unexplored chemical space. We herein report a visible light-driven approach to these compounds that relies on an intramolecular crossed [2 + 2] photocycloaddition of styrene derivatives enabled by triplet energy transfer. Bicyclo[2.1.1]hexanes were obtained in good to high yields (19 examples, 61%-quantitative yield) and allowed for further functionalizations by consecutive reactions, thereby opening different pathways to decorate the aliphatic core structure.
Overview
From T. Rigotti and T. Bach. Who needs the RCM when you can bring two alkenes together in a light catalyzed 2+2 cyclization. It would be nice to see if this translates to larger ring size [1.1.n]- bicyclics.
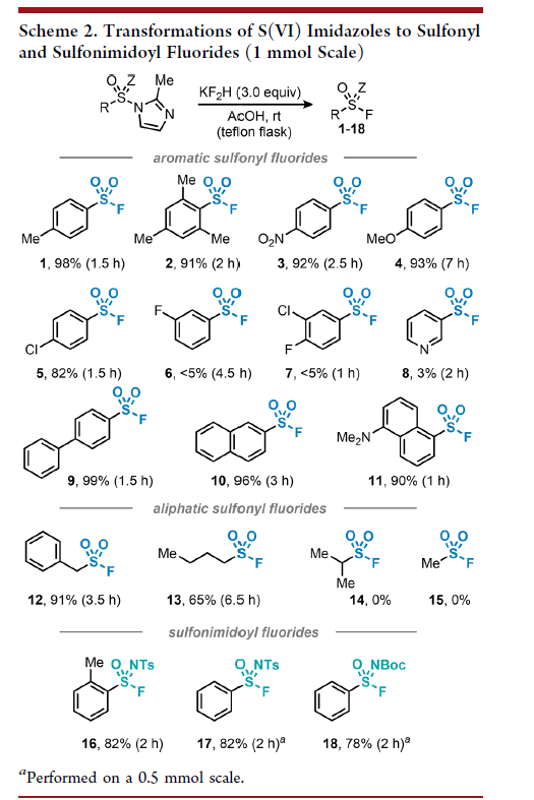
Acid-Mediated Imidazole-to-Fluorine Exchange for the Synthesis of Sulfonyl and Sulfonimidoyl Fluorides
Authors: Marco T. Passia, Joachim Demaerel, Mostafa M. Amer, Alwin Drichel, Stefanie Zimmer, and Carsten Bolm
Abstract
Sulfur(VI) fluoride motifs are important entities in organic chemistry. Typically, their syntheses involve the corresponding chlorides, which are often difficult to prepare and characterized by a poor storability due to the inherently weak S–Cl bond. Here, a single-step procedure for the preparation of sulfur(VI) fluorides starting from sulfonyl imidazoles as stable S(VI) reservoirs is described. By using a simple combination of AcOH and potassium bifluoride (KF2H), an imidazole-to-fluorine exchange furnishes a variety of sulfonyl, sulfonimidoyl, sulfoxyl, and sulfamoyl fluorides in good to excellent yields.
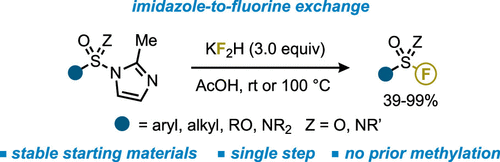
Overview
From M. T. Passia, C. Bolm et al. Sulfonyl-F compounds are useful sulfonation reagents. Here is a method which works also for sulfonimides.