This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

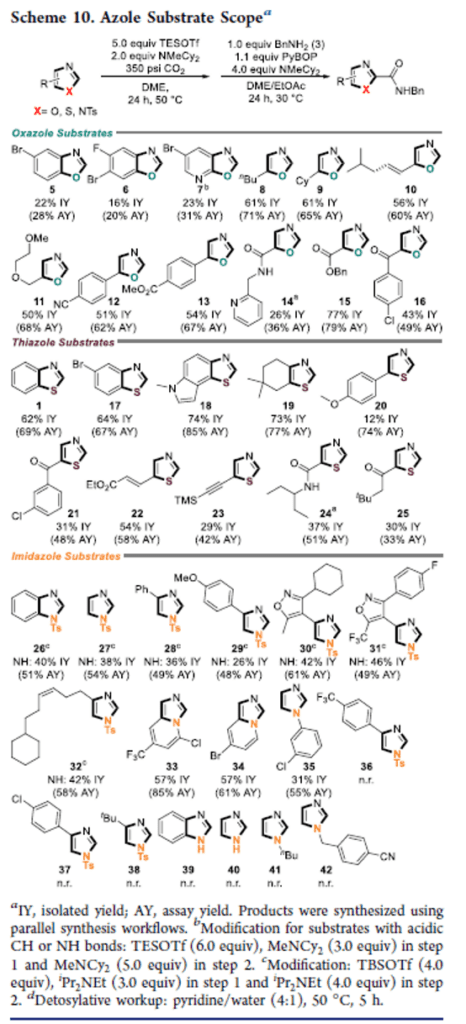
Accessing Diverse Azole Carboxylic Acid Building Blocks via Mild C–H Carboxylation: Parallel, One-Pot Amide Couplings and Machine-Learning-Guided Substrate Scope Design
Authors: Stephanie Felten, Cyndi Qixin He, Mark Weisel, Michael Shevlin, and Marion H. Emmert
Abstract
This manuscript describes a mild, functional group tolerant, and metal-free C–H carboxylation that enables direct access to azole-2-carboxylic acids, followed by amide coupling in one pot. This demonstrates a significant expansion of the accessible chemical space of azole-2-amides, compared to previously known methodologies. Key to the described reactivity is the use of silyl triflate reagents, which serve as reaction mediators in C–H deprotonation and stabilizers of (otherwise unstable) azole carboxylic acid intermediates. A diverse azole substrate scope designed via machine-learning-guided analysis demonstrates the broad utility of the sequence. Density functional theory calculations provide detailed insights into the role of silyl triflates in the reaction mechanism. Transferrable applications of the protocol are successfully established: (i) A low pressure (CO2 balloon) option for synthesizing azole-2-carboxylic acids without the need for high-pressure equipment; (ii) the use of 13CO2 for the synthesis of labeled compounds; (iii) isocyanates as alternative electrophiles for direct C–H amidation; (iv) and the use of the developed chemistry in a 24 × 12 parallel synthesis workflow with a 90% library success rate. Fundamentally, the reported protocol expands the use of heterocycle C–H functionalization from late-stage functionalization applications toward its use in library synthesis. It provides general access to densely functionalized azole-2-carboxylic acid building blocks and demonstrates their one-pot diversification.
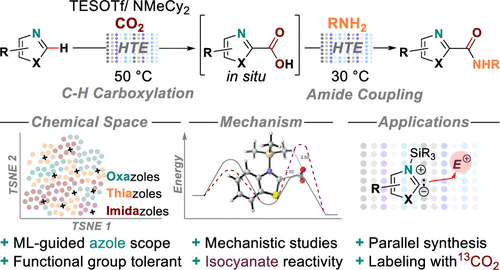
Overview
From S. Felten, C. Q. He, M. H. Emmert et al. A relatively mild method to make 2-Carboxy-azoles and their amides as long as you can make 350 psi CO2. To be fair, the reaction works with balloon pressure CO2 albeit in slightly decreased yield. There is a fair amount of multidimensional optimization and mechanistic proof work. Here is an example table:

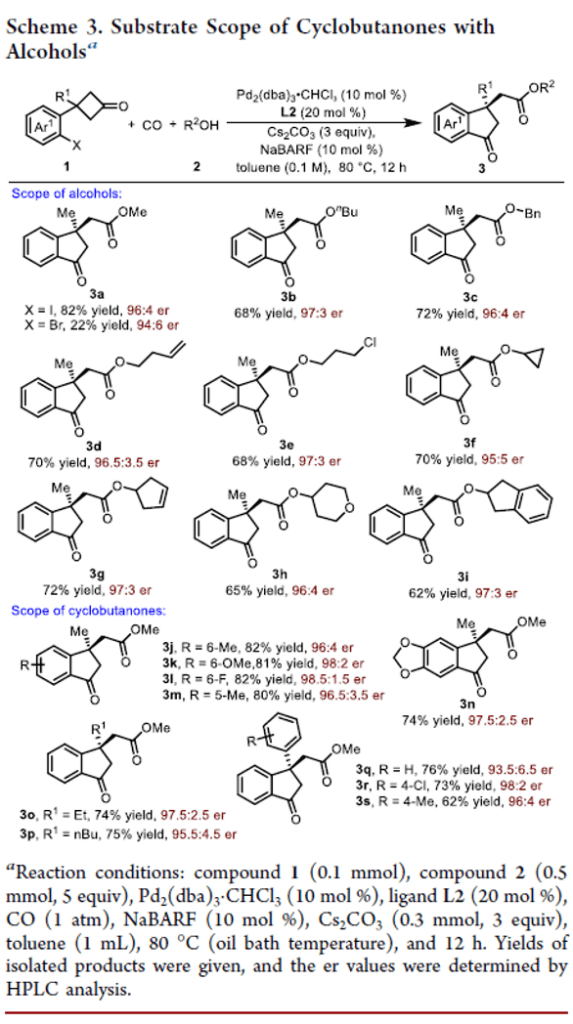
Palladium-Catalyzed Asymmetric C–C Bond Activation/Carbonylation of Cyclobutanones
Authors: Long Chen, Cong Shi, Wei Li, Bo Li, Jie Zhu, Aijun Lin, and Hequan Yao
Abstract
A palladium-catalyzed asymmetric C–C bond activation/carbonylation of cyclobutanones with CO has been developed. This reaction provided an efficient method for the synthesis of chiral indanones bearing a quaternary carbon stereocenter in good yields with an excellent enantiomeric ratio, exhibiting good functional group tolerance. Transformations of the products to chiral 3,4-dihydroquinolin-2(1H)-one and 1H-indene further demonstrated the versatility of this reaction.
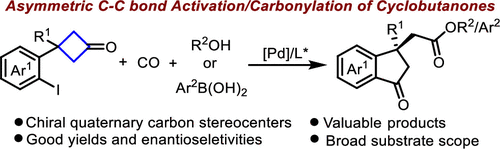
Overview
From L. Chem, A. Lin, H. Yao et al. Here is a Pd-catalyzed ring expansion of a cyclobutanone with CO mediated CO2R formation. Tricky!
See the following table:

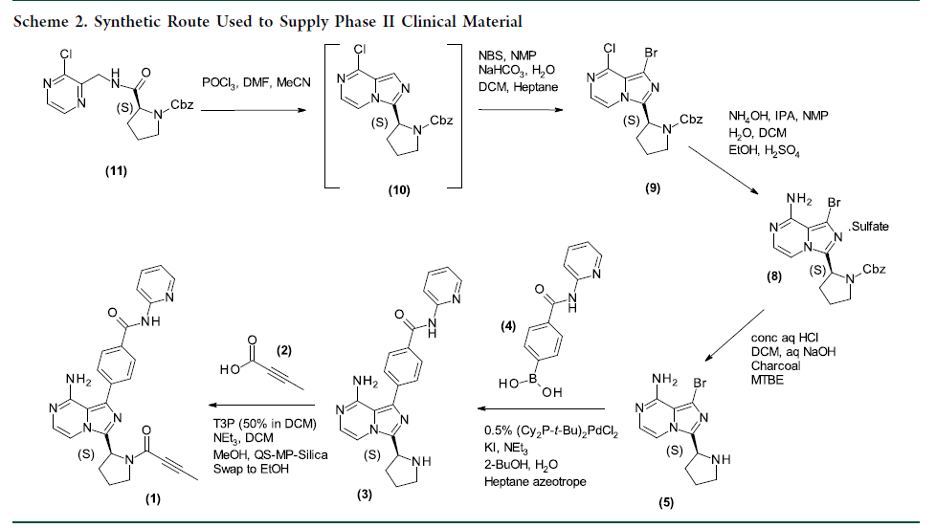
Development of Commercial Manufacturing Processes for Acalabrutinib
Authors: Paul A. Bethel, Lai Chun Chan, Katie Cooper, Gareth J. Ensor, Jerry Evarts, Rustam Garrey, J. Gair Ford, Daniel Harding, Shaun Hughes, Lucy Jackson, Michael Golden, Kirsty Millard, Andrew Phillips, Andrew Robbins, David Short, Alex Telford, and David T. E. Whittaker
Abstract
The development of processes to produce the Bruton tyrosine kinase inhibitor, acalabrutinib 1, has resulted in improvements to the yield, cycle time, and operability, to realize a robust commercial manufacturing process. A highly accelerated clinical program meant that numerous key process challenges had to be resolved in a short timeframe. Issues are discussed, such as the uncontrolled epimerization of a chiral center and the control of the acalabrutinib 1 crystallization step, which was prone to oiling. Specifically, work to understand the complex polymorph landscape of a key intermediate (to facilitate resolution of a filtration issue) is described.
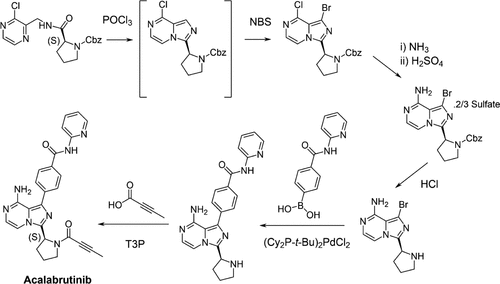
Overview
From P. A. Bethel, M. Golden et al. A process scaled imidazopyrazine drug synthesis. Here is an early process route. Other useful methods are in there as well.

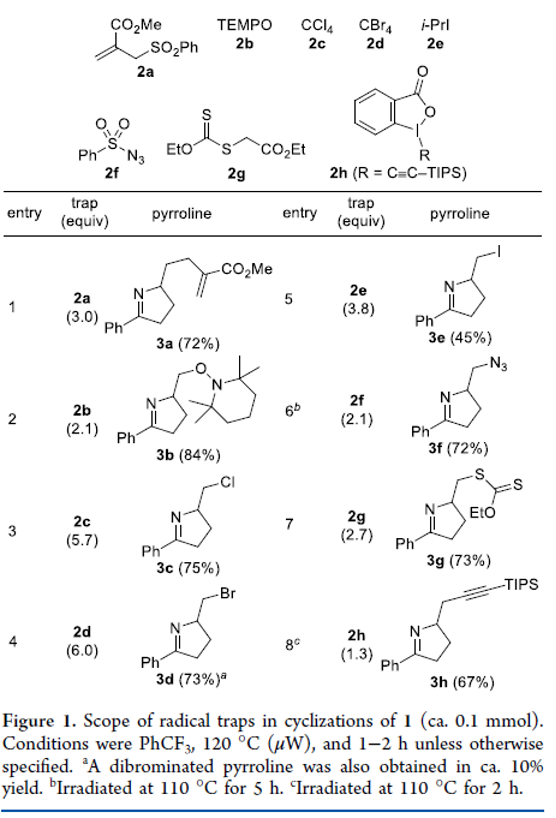
Microwave- and Thermally Promoted Iminyl Radical Cyclizations: A Versatile Method for the Synthesis of Functionalized Pyrrolines
Authors: Jatinder Singh, Tanner J. Nelson, Samuel A. Mansfield, Garrison A. Nickel, Yu Cai, Dakota D. Jones, Jeshurun E. Small, Daniel H. Ess, and Steven L. Castle
A detailed study of iminyl radical cyclizations of O-aryloximes tethered to alkenes is reported. The reactions can be triggered by either microwave irradiation or conventional heating in an oil bath. A variety of radical traps can be employed, enabling C–C, C–N, C–O, C–S, or C–X bond formation and producing a diverse array of functionalized pyrrolines. Substrates containing an allylic sulfide furnish terminal alkenes by a tandem cyclization–thiyl radical β-elimination pathway. Cyclizations of hydroxylated substrates exhibit moderate diastereoselectivity that in some cases can partially be attributed to intramolecular hydrogen bonding. Computational studies suggested a possible role for thermodynamics in controlling the stereochemistry of cyclizations. The reaction temperature can be lowered from 120 to 100 °C by employing O-(p–tert-butylphenyl)oximes instead of O-phenyloximes as substrates, and these second-generation iminyl radical precursors can be used in a one-pot oxime ether formation–cyclization that is promoted by conventional heating. The functionalized pyrrolines obtained from these reactions can be conveniently transformed in several different ways.
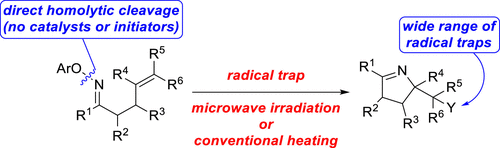
Overview
From J. Singh, S. L. Castle et al. How to generate an imminyl radical, cyclize it into a pi-system and cap that with a radical trap. Examples: