This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

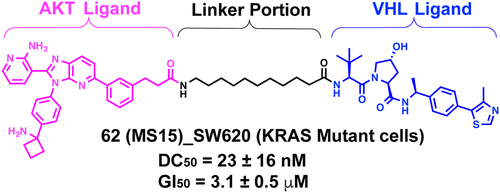
Novel Allosteric Inhibitor-Derived AKT Proteolysis Targeting Chimeras (PROTACs) Enable Potent and Selective AKT Degradation in KRAS/BRAF Mutant Cellsoles via Oxidative Cyclization of N-Tosylhydrazones and Anilines
Authors: Xufen Yu, Jia Xu, Kaitlyn M. Cahuzac, Ling Xie, Yudao Shen, Xian Chen, Jing Liu, Ramon E. Parsons, Jian Jin
Abstract
AKT is an important target for cancer therapeutics. Significant advancements have been made in developing ATP-competitive and allosteric AKT inhibitors. Recently, several AKT proteolysis targeting chimeras (PROTACs) derived from ATP-competitive AKT inhibitors have been reported, including MS21. While MS21 potently degraded AKT and inhibited the growth in tumor cells harboring PI3K/PTEN pathway mutation, it was largely ineffective in degrading AKT in KRAS/BRAF mutated cells as a single agent. To overcome the AKT degradation resistance in KRAS/BRAF mutated cells, we developed novel AKT PROTACs derived from an AKT allosteric inhibitor, including degrader 62 (MS15). 62 displayed potent and selective AKT degradation activity and potent antiproliferative activity in KRAS/BRAF mutated cancer cells, in addition to PI3K/PTEN mutated cancer cells. Furthermore, 62 was bioavailable in mice through intraperitoneal administration. Overall, 62 is a valuable chemical tool to degrade AKT in cells harboring KRAS/BRAF mutation and expands the tool box for pharmacologically modulating AKT.
Overview
From the J. Liu, R. E. Parsons and J. Jin groups. This PROTAC paper presents a comprehensive linker examination connecting one AKT allosteric inhibitor to 2 different VHL ligands. About 60 PEG and alkyl linkers with amide connections at each end were investigated. The PROTAC degraded AKT in KRAS/BRAF mutated cells as a major advance.

Discovery of 5,7-Dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-ones as Highly Selective CDK2 Inhibitors
Authors: Alexander Sokolsky, Sarah Winterton, Keith Kennedy, Katherine Drake, Kristine Stump, Lu Huo, Yvonne Lo, Min Ye, Maryanne Covington, Sharon Diamond, Yan-ou Yang, Sunkyu Kim, Swamy Yeleswaram, Liangxing Wu, Wenqing Yao
Abstract
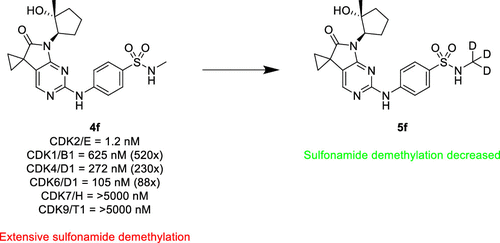
A series of exceptionally selective CDK2 inhibitors are described. Starting from an HTS hit, we successfully scaffold hopped to a 5,7-dihydro-6H-pyrrolo[2,3-d]pyrimidin-6-one core structure, which imparted a promising initial selectivity within the CDK family. Extensive further SAR identified additional factors that drove selectivity to above 200× for CDKs 1/4/6/7/9. General kinome selectivity was also greatly improved. Finally, use of in vivo metabolite identification allowed us to pinpoint sulfonamide dealkylation as the primary metabolite, which was ameliorated through the deuterium effect.
Overview
From the A. Sokolsky group. 1 of 2 papers released on the same day which found success in changing a SO2NHCH3 to a SO2NHCD3. Good to keep an eye on the deuterium approach to stabilize small molecules against in vivo metabolism.

Approaches to Synthesis and Isolation of Enantiomerically Pure Biologically Active Atropisomers
Authors: Anna-Carin C. Carlsson, Staffan Karlsson, Rachel H. Munday, Matthew R. Tatton
Abstract
Conspectus
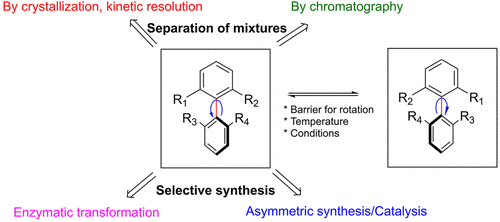
Atropisomerism is a stereochemical phenomenon exhibited by molecules containing a rotationally restricted σ bond. Contrary to classical point chirality, the two atropisomeric stereoisomers exist as a dynamic mixture and can be interconverted without the requirement of breaking and reforming a bond. Although this feature increases structural complexity, atropisomers have become frequent targets in medicinal chemistry projects. Their axial chirality, e.g., from axially chiral biaryl motifs, gives access to unique 3D structures. It is often desirable to have access to both enantiomers of the atropisomers via a nonselective reaction during the early discovery phase as it allows the medicinal chemistry team to probe the structure activity relationship in both directions. However, once a single atropisomer is selected, it presents several problems. First, the pure single atropisomer may interconvert to the undesired stereoisomer under certain conditions. Second, separation of atropisomers is nontrivial and often requires expensive chiral stationary phases using chromatography or additives if a salt resolution approach is chosen. Other options can be kinetic resolution using enzymes or chiral catalysts. However, apart from the high cost often associated with the two latter methods, a maximum yield of only 50% of the desired atropisomer can be obtained. The ideal approach is to install the chiral atropisomeric axis enantioselectively or employing a dynamic kinetic resolution approach. In theory, both approaches have the potential to provide a single atropisomer in quantitative yield. This Account will discuss the successes/failures and challenges we have experienced in developing methods for resolution/separation and asymmetric synthesis of atropisomeric drug candidates in one of our early phase drug development projects. Suitability for the different methods at various stages of the drug development phase is discussed. Depending on the scale and time available, a separation of a mixture of atropisomers by chromatography was sometimes preferred, whereas asymmetric- or resolution approaches were desired for long-term supply. With the use of chromatography, the impact on separation efficiency and solvent consumption, depending on the nature of the substrate, is discussed. We hope that with this Account the readers will get a better view on the challenges medicinal and process chemists meet when designing new atropisomeric drug candidates and developing processes for manufacture of a single atropisomer.
Overview
From the Carlsson and Karlsson group. Another topical review of how to resolve biaryl-atropisomers. This is a current popular area of novel small molecule drug discovery to be aware of.

Synthesis of Imidized Cyclobutene Derivatives by Strain Release of [1.1.1]Propellane
Authors: Xiaofan Du, Di Xu, Gongcheng Xu, Chuanming Yu, Xinpeng Jiang
Abstract
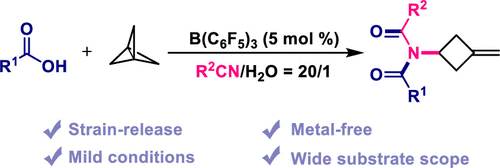
Herein, we report the metal-free synthesis of imidized methylene cyclobutane derivatives via a strain-release driven addition reaction of [1.1.1]propellane. Using this strategy, the methylene cyclobutyl cation intermediate generated by protonation of [1.1.1]propellane was found to be trapped by nitriles to form a nitrilium ion intermediate, which subsequently reacted with carboxylic acids to produce imidized methylene cyclobutene derivatives via a Mumm-type rearrangement.
Overview
From the X. Jiang group. I am not quite sure how this will be useful in the future but I like BCP chemistry and I like finding ways to access cyclobutanes. The square shaped molecule has lots of untapped potential in medicinal chemistry. Now if we could just get a cheap, safe supply of [1.1.1]propellane!
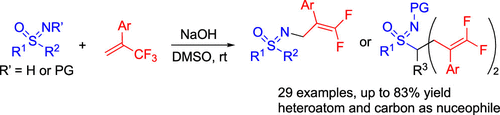
Superbase-Mediated gem-Difluoroalkenylations of Sulfoximines
Authors: Xianliang Wang, Chenyang Wang, Carsten Bolm
Abstract
At ambient temperature, deprotonated sulfoximines react with 1-trifluoromethylalkenes to provide either N- or C-gem-difluoroalkenylated products. The reaction site depends upon the N substituent of the starting material. The optimal conditions involve the use of a superbasic system NaOH in dimethyl sulfoxide. The reactions are characterized by a broad substrate scope and medium to high yields. Scale-up experiments of both the N- and C-gem-difluoroalkenylations proceeded well. Treatment of a N-difluoroallyl sulfoximine with an aryl thiol under dioxygen afforded the corresponding oxygenated addition product.
Overview
From the C. Bolm group. Bolm is a pioneer in the sulfoximine area. Here he connects two moieties that medicinal chemists find very stylish, sulfoximines and difluoroalkenes. This could lead to something new.
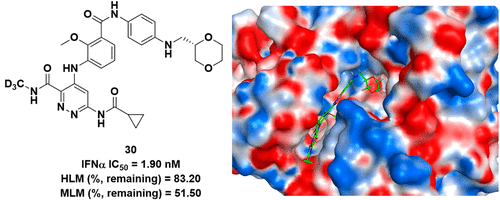
Novel TYK2 Inhibitors with an N-(Methyl-d3)pyridazine-3-carboxamide Skeleton for the Treatment of Autoimmune Diseases
Authors: Fei Liu, Bin Wang, Yanlong Liu, Wei Shi, Xujing Tang, Xiaojin Wang, Zhongyuan Hu, Ying Zhang, Yahui Guo, Xiayun Chang, Xiangyi He, Hongjiang Xu, Ying He
Abstract
Tyrosine kinase 2 (TYK2) mediates the interleukin-23 (IL-23), IL-12, and type I interferon (IFN)-driven signal responses that are critical in autoimmune diseases. Here, a series of novel derivatives with an N-(methyl-d3)pyridazine-3-carboxamide skeleton that bind to the TYK2 pseudokinase domain were designed, synthesized, and evaluated. Among them, compound 30 demonstrated more excellent inhibitory potency against STAT3 phosphorylation than the positive control deucravacitinib. In addition to JAK isoform selectivity, compound 30 exhibited good in vivo and in vitro pharmacokinetic properties. Furthermore, compound 30 was orally highly effective in both IL-23-driven acanthosis and anti-CD40-induced colitis models. Together, these findings support compound 30 as a promising candidate for therapeutic applications in autoimmune diseases.
Overview
From the Y. He group. The 2nd of 2 papers released on the same day to highlight the benefits of changing a CH3 to a CD3. This one has extensive use of the CD3NH-amide in many analogues.



