This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

Bicyclobutanes: from curiosities to versatile reagents and covalent warheads
Authors: Christopher B. Kelly, John A. Milligan, Leon J. Tilley and Taylor M. Sodano
Abstract
The unique chemistry of small, strained carbocyclic systems has long captivated organic chemists from a theoretical and fundamental standpoint. A resurgence of interest in strained carbocyclic species has been prompted by their potential as bioisosteres, high fraction of sp3 carbons, and limited appearance in the patent literature. Among strained ring systems, bicyclo[1.1.0]butane (BCB) stands apart as the smallest bicyclic carbocycle and is amongst the most strained carbocycles known. Despite the fact that BCBs have been synthesized and studied for well over 50 years, they have long been regarded as laboratory curiosities. However, new approaches for preparing, functionalizing, and using BCBs in “strain-release” transformations have positioned BCBs to be powerful synthetic workhorses. Further, the olefinic character of the bridgehead bond enables BCBs to be elaborated into various other ring systems and function as covalent warheads for bioconjugation. This review will discuss the recent developments in the synthesis and functionalization of BCBs as well as the applications of these strained rings in synthesis and drug discovery. An overview of the properties and the historical context of this interesting structure will be provided.

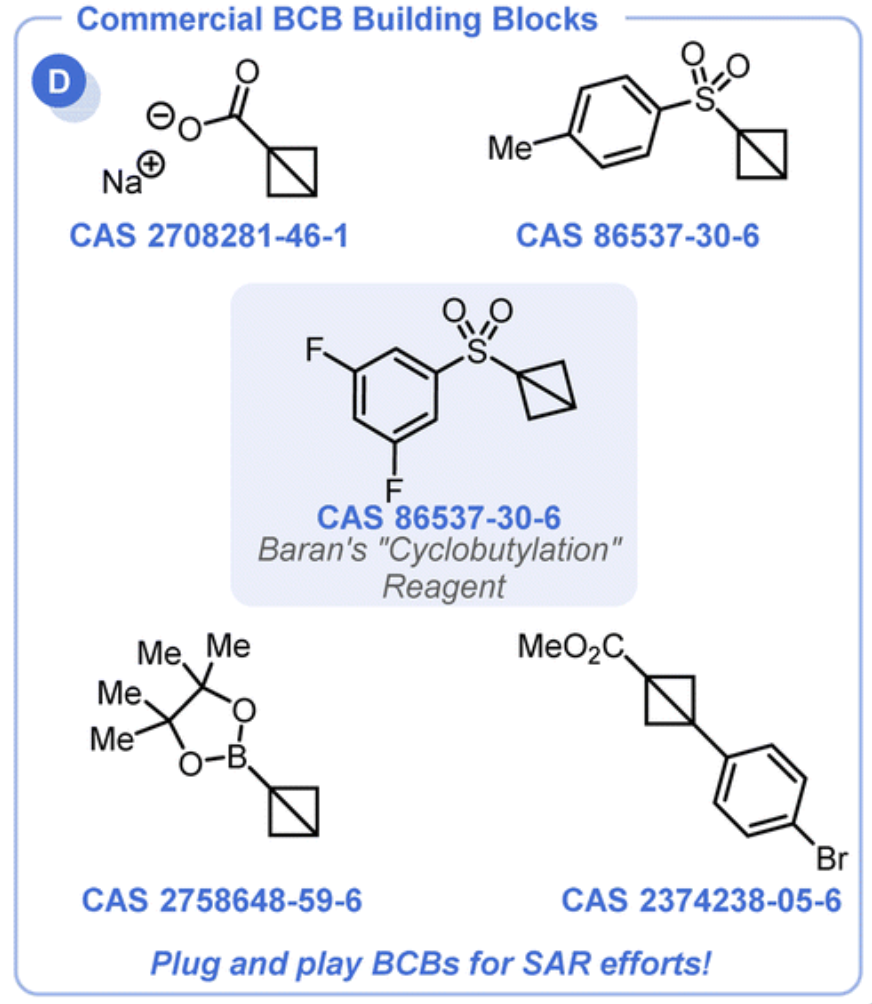
Overview
From the C. B. Kelly group. Chem. Sci. 2022, 13, 11721-11737. This is a review on bicyclobutanes (BCBs). The parent is made of 2 fused cyclopropanes. These molecules are just becoming commercially available but are still quite expensive with the cheapest (the Baran reagent) at about $800/1g. With time this price should drop. Note the Baran reagent is 1862202-13-8. The reagent 2708281-46-1 works as a covalent reactive group. Phenylacrylamide GSH T1/2 = 1.2 h while Phenyl-NH-BCB-amide GSH T1/2 = 33h.
Recent Advances in Alkenyl sp2 C–H and C–F Bond Functionalizations: Scope, Mechanism, and Applications
Authors: Ming-Zhu Lu, Jeffrey Goh, Manikantha Maraswami, Zhenhua Jia, Jie-Sheng Tian, Teck-Peng Loh
Abstract
Alkenes and their derivatives are featured widely in a variety of natural products, pharmaceuticals, and advanced materials. Significant efforts have been made toward the development of new and practical methods to access this important class of compounds by selectively activating the alkenyl C(sp2)–H bonds in recent years. In this comprehensive review, we describe the state-of-the-art strategies for the direct functionalization of alkenyl sp2 C–H and C–F bonds until June 2022. Moreover, metal-free, photoredox, and electrochemical strategies are also covered. For clarity, this review has been divided into two parts; the first part focuses on currently available alkenyl sp2 C–H functionalization methods using different alkene derivatives as the starting materials, and the second part describes the alkenyl sp2 C–F bond functionalization using easily accessible gem-difluoroalkenes as the starting material. This review includes the scope, limitations, mechanistic studies, stereoselective control (using directing groups as well as metal-migration strategies), and their applications to complex molecule synthesis where appropriate. Overall, this comprehensive review aims to document the considerable advancements, current status, and emerging work by critically summarizing the contributions of researchers working in this fascinating area and is expected to stimulate novel, innovative, and broadly applicable strategies for alkenyl sp2 C–H and C–F bond functionalizations in the coming years.
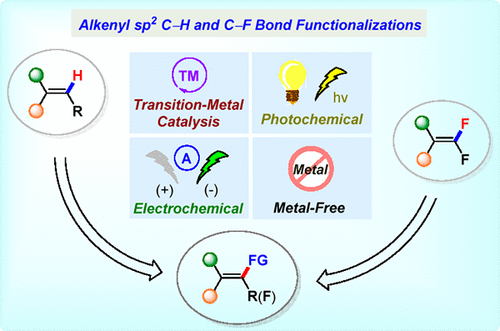
Overview
From the T.-P. Loh group. This is a 167-page review of alkene-CH activation/substitution reactions with the added bonus of a review of nucleophilic additions onto geminal alkenyl difluorides. This is a useful addition to your library for sure!

Acyclic and Heterocyclic Azadiene Diels–Alder Reactions Promoted by Perfluoroalcohol Solvent Hydrogen Bonding: Comprehensive Examination of Scope
Authors: Zixi Zhu, Dale L. Boger
Herein, the first use of perfluoroalcohol H-bonding in accelerating acyclic azadiene inverse electron demand cycloaddition reactions is described, and its use in the promotion of heterocyclic azadiene cycloaddition reactions is generalized through examination of a complete range of azadienes. The scope of dienophiles was comprehensively explored; relative reactivity trends and solvent compatibilities were established with respect to the dienophile as well as azadiene; H-bonding solvent effects that lead to rate enhancements, yield improvements, and their impact on regioselectivity and mode of cycloaddition are defined; new viable diene/dienophile reaction partners in the cycloaddition reactions are disclosed; and key comparison rate constants are reported. The perfluoroalcohol effectiveness at accelerating an inverse electron demand Diels–Alder cycloaddition is directly correlated with its H-bond potential (pKa). Not only are the reactions of electron-rich dienophiles accelerated but those of strained and even unactivated alkenes and alkynes are improved, including representative bioorthogonal click reactions.
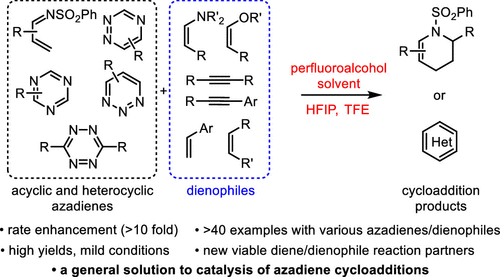
Overview
From the D. Boger group. This new addition to his inverse demand Diels-Alder reaction methodology explores the benefit of accelerating these reactions in fluorinated alcohol solvents. Trifluoroethanol (TFE, pKa = 12.4) and hexafluoroisopropanol (HFIP, pKa = 9.3) were particularly useful due to pKa driven H-bonding effects with substrates in transition states and their additional non-nucleophilic nature from steric hinderance. Rings made include pyridazines, 1,2,4-triazenes, unusual pyridines, and pyrimidines.

Discovery of Chlorofluoroacetamide-Based Covalent Inhibitors for Severe Acute Respiratory Syndrome Coronavirus 2 3CL Protease
Authors: Yuya Hirose, Naoya Shindo, Makiko Mori, Satsuki Onitsuka, Hikaru Isogai, Rui Hamada, Tadanari Hiramoto, Jinta Ochi, Daisuke Takahashi, Tadashi UedaJose M. M. Caaveiro, Yuya Yoshida, Shigehiro Ohdo, Naoya Matsunaga, Shinsuke Toba, Michihito Sasaki, Yasuko Orba, Hirofumi Sawa, Akihiko Sato, Eiji Kawanishi, Akio Ojida
Abstract
The coronavirus disease 2019 (COVID-19) pandemic has necessitated the development of antiviral agents against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). 3C-like protease (3CLpro) is a promising target for COVID-19 treatment. Here, we report a new class of covalent inhibitors of 3CLpro that possess chlorofluoroacetamide (CFA) as a cysteine-reactive warhead. Based on an aza-peptide scaffold, we synthesized a series of CFA derivatives in enantiopure form and evaluated their biochemical efficiency. The data revealed that 8a (YH-6) with the R configuration at the CFA unit strongly blocks SARS-CoV-2 replication in infected cells, and its potency is comparable to that of nirmatrelvir. X-ray structural analysis showed that YH-6 formed a covalent bond with Cys145 at the catalytic center of 3CLpro. The strong antiviral activity and favorable pharmacokinetic properties of YH-6 suggest its potential as a lead compound for the treatment of COVID-19.
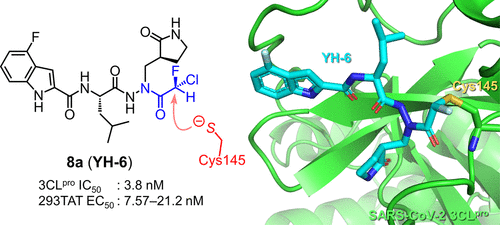
Overview
From the E. Kawanishi and A. Ojida groups. Targeting the Cys145 of coronavirus 2 3CL Protease, a chlorofluoroacetamide (CFA) warhead proved very effective at selectively (vs. other key kinases) forming a covalent bond to this enzyme. The Cl-atom is displaced. It also showed little reactivity to GSH, T1/2 = 48.2h. Finally the CFA-thiol adduct is readily hydrolyzed from the covalent connection which eliminates solvent-exposed off-target labeling. It had a promising PK-profile, 100 mpk (mouse) showed 15.2 µM after 1h and 0.4 µM after 6h, yet with i.v. comparison the F = 9%. This CFA is an interesting warhead considering that chloroacetamide is considered much too reactive to be useful as a covalent inhibitor warhead.

Development of a C–C Bond Cleavage/Vinylation/Mizoroki–Heck Cascade Reaction: Application to the Total Synthesis of 14- and 15-Hydroxypatchoulol
Authors: Christina G. Na, Suh Hyun Kang, Richmond Sarpong
Abstract
A C–C bond cleavage/vinylation/Mizoroki–Heck cascade reaction has been developed to provide access to densely functionalized bicyclo[2.2.2]octane frameworks. The sequence proceeds through the coupling of dihydroxylated pinene derivatives, prepared from carvone, with gem-dichloroalkenes. The method was applied to 12-step total syntheses of both 14- and 15-hydroxypatchoulol, which provided unambiguous support for the structure of the natural products and corrects a misassignment in the isolation report.
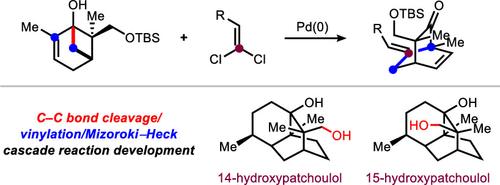
Overview
From the R. Sarpong group. This one might be a little difficult to imagine applying in a medchem sense but it is interesting to see how modern sesquiterpene total synthesis is being done. Here is the key reaction:
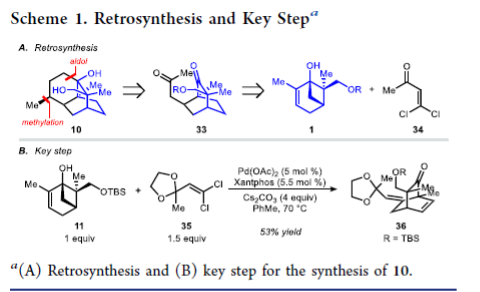
A bridgehead -OH group on a 3.1.1-bicycloheptane is opened under Pd catalysis and reacted with a dichlorovinyl group to make a new bicyclic ring. It is a level of creativity fitting for a JACS publication. Other reactions employed are interesting to read also.

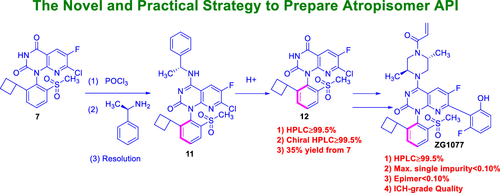
Process Research for ZG1077, a KRAS G12C Inhibitor
Authors: Wei-Dong Feng, Bin-Hua Lv, Wei Ye, Cai Wang, Lin-Yong Jin, Run-Qing Wang, Chang-Min Lian, Kuan-Hong You
Abstract
A novel, efficient, and robust synthetic route to ZG1077 with an atropisomer structure for KRAS G12C inhibitors was designed. The critical process parameters were optimized and established to reduce or avoid process-related impurities. The formation mechanism, purge pathways, and control strategy for these impurities are discussed. Compared with the medicinal chemistry route, the single atropisomer drug substance was obtained with chiral resolution rather than the supercritical fluid chromatogram purification technique, and it was obtained in 3.01% overall yield with >99.5% purity and 99.8% e.e. in the novel route. However, the overall yield is only 0.56%, and the purity and chiral purity were less than 99.0% in the medicinal chemistry route. The process is suitable to obtain enough active pharmaceutical ingredients for preclinical and clinical studies.
Overview
From the W.-D. Feng and B.-H. Lv group. The process chemistry involved for the clinical supply synthesis of the covalent inhibitor of KRAS G12C, ZG1077. This is an atropisomer and it is resolved by selective crystallization from refluxing acetone or THF.

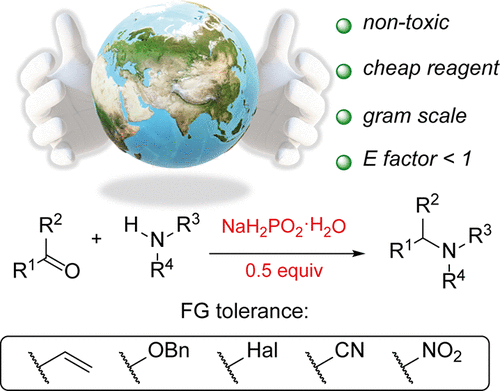
Sodium Hypophosphite as a Bulk and Environmentally Friendly Reducing Agent in the Reductive Amination
Authors: Wei-Dong Feng, Bin-Hua Lv, Wei Ye, Cai Wang, Lin-Yong Jin, Run-Qing Wang, Chang-Min Lian, Kuan-Hong You
Abstract
NaH2PO2 was found to promote reductive amination. Being nontoxic, stable, environmentally benign, and available in bulk amounts, this reducing agent showed a powerful potential to compete with classical reductants applied in the target process. An E factor of 1 was achieved for the substrate scope. Different carbonyl compounds reacted with amines under the developed conditions. The reaction demonstrated a great compatibility with a wide range of functional groups. Reaction conditions were scaled up to 200-fold.
Overview
From the D. Chusov group. Use of NaH2PO2 as an alternative to STAB and other reagents for reductive amination reactions. Hmmmmm.



