This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

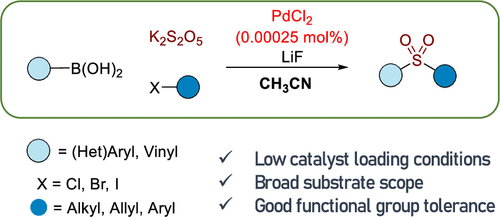
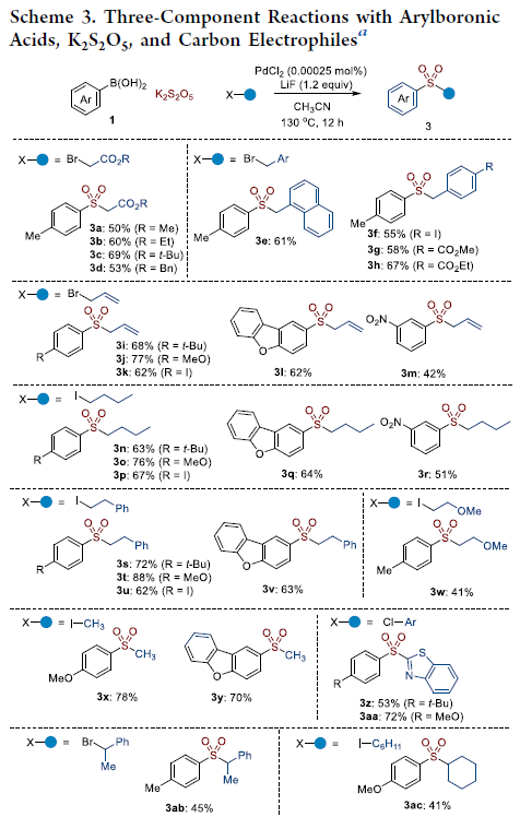
Highly Reactive Palladium-Catalyzed and Acetonitrile-Mediated Three-Component Reactions for Arylsulfone Synthesis
Authors: Muhammad Aliyu Idris, Sunwoo Lee
Abstract
Arylsulfone groups play an important role in the synthesis of functionalized molecules. The acetonitrile-mediated three-component reactions for arylsulfone synthesis were developed in the presence of a 0.00025 mol % palladium catalyst. Arylboronic acids reacted with potassium metabisulfite (K2S2O5) and benzyl bromide in the presence of LiF and a very low concentration of PdCl2 in acetonitrile solvents to produce the corresponding benzyl arylsulfones in moderate to good yields. Various arylboronic acids reacted with K2S2O5 and carbon electrophiles to produce the desired arylsulfones under the optimized conditions. It was proposed that acetonitrile accelerated the generation of aryl anion species from the reaction of arylboronic acids and LiF.
Overview
From M. A. Idris and S. Lee. A simple way to make arylsulfones with almost anything on the other side.

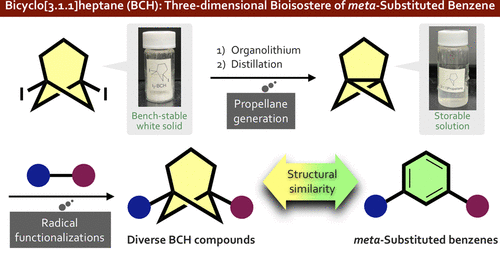
Practical and Facile Access to Bicyclo[3.1.1]heptanes: Potent Bioisosteres of meta-Substituted Benzenes
Authors: Toranosuke Iida, Junichiro Kanazawa, Tadafumi Matsunaga, Kazunori Miyamoto, Keiichi Hirano, Masanobu Uchiyama
Abstract
There is increasing interest in replacement of the planar aromatic rings of drug candidates with three-dimensional caged scaffolds in order to improve the physical properties, but bioisosteres of meta-substituted benzenes have remained elusive. We focused on the bicyclo[3.1.1]heptane (BCH) scaffold as a novel bioisostere of meta-substituted benzenes, anticipating that [3.1.1]propellane (2) would be a versatile precursor of diversely functionalized BCHs. Here, we describe a practical preparative method for [3.1.1]propellane from newly developed 1,5-diiodobicyclo[3.1.1]heptane (1), as well as difunctionalization reactions of 2 leading to functionalized BCHs. We also report postfunctionalization reactions of these products.
Overview
From T.Iida, J. Kanazawa, K. Hirano, M. Uchiyama et al. With reactivity essentially the same as [1.1.1]propellane, [3.1.1]propellane offers an sp3 entry to meta-phenyl substituted analogues with BCH derivatives. The diiodo-precursor of [3.1.1]propellane is easy to make and stable yet because of the reactivity, [3.1.1] needs to be co-distilled in TBME at 0C and stored at -30C.
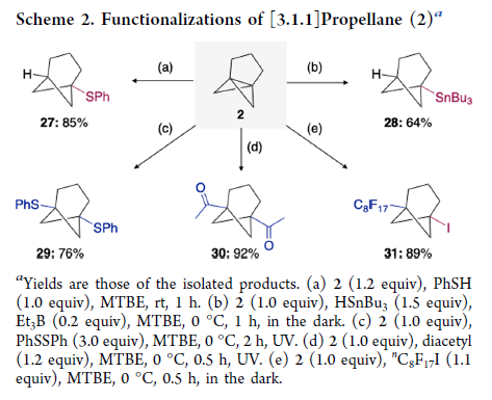
In case you are interested in familial comparisons of reactivity, this information is in the SI:


Structural Basis for Inhibition of Mutant EGFR with Lazertinib (YH25448)
Authors: David E. Heppner, Florian Wittlinger, Tyler S. Beyett, Tatiana Shaurova, Daniel A. Urul, Brian Buckley, Calvin D. PhamIlse K. Schaeffner, Bo Yang, Blessing C. Ogboo, Earl W. May, Erik M. Schaefer, Michael J. EckStefan A. Laufer, Pamela A. Hershberger
Abstract
Lazertinib (YH25448) is a novel third-generation tyrosine kinase inhibitor (TKI) developed as a treatment for EGFR mutant non-small cell lung cancer. To better understand the nature of lazertinib inhibition, we determined crystal structures of lazertinib in complex with both WT and mutant EGFR and compared its binding mode to that of structurally related EGFR TKIs. We observe that lazertinib binds EGFR with a distinctive pyrazole moiety enabling hydrogen bonds and van der Waals interactions facilitated through hydrophilic amine and hydrophobic phenyl groups, respectively. Biochemical assays and cell studies confirm that lazertinib effectively targets EGFR(L858R/T790M) and to a lesser extent HER2. The molecular basis for lazertinib inhibition of EGFR reported here highlights previously unexplored binding interactions leading to improved medicinal chemistry properties compared to clinically approved osimertinib (AZD9291) and offers novel strategies for structure-guided design of tyrosine kinase inhibitors.
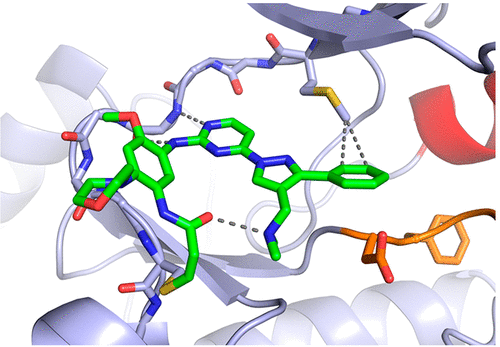
Overview
From D. E Heppner et al. Structural basis for EGFR inhibitory series just begs for macrocyclic analogues to be made!
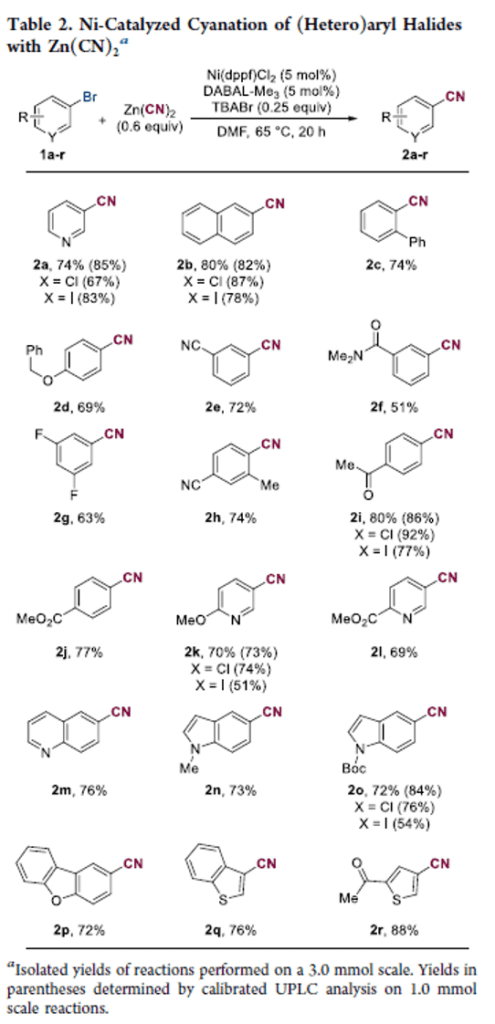
Nickel-Catalyzed Cyanation of (Hetero)aryl Bromides Using DABAL-Me3 as a Soluble Reductant
Authors: Geraldo Duran-Camacho, J. Caleb Hethcox
Abstract
A nickel-catalyzed cyanation of (hetero)aryl halides using air-stable bis(trimethylaluminum)-1,4-diazabicyclo[2.2.2]octane (DABAL-Me3) as a soluble reductant has been developed. The reaction uses readily available and inexpensive Ni(dppf)Cl2 as a precatalyst, a substoichiometric amount of Zn(CN)2, and DABAL-Me3 as an alternative to commonly prescribed insoluble reductants. We found the addition of catalytic tetrabutylammonium bromide (TBABr) to be beneficial, due to facilitating dissolution of low levels of the cyanide salt (confirmed via a control study). Similarly, slow addition of a tetrabutylammonium cyanide (TBACN) solution is effective and results in a completely homogeneous reaction mixture.

Overview
From G. Duran-Camacho and J. C. Hethcox. Mile cyanation reactions are always useful.

Discovery of D6808, a Highly Selective and Potent Macrocyclic c-Met Inhibitor for Gastric Cancer Harboring MET Gene Alteration Treatment
Authors: Chaofan Wang, Jie Li, Lingzhi Qu, Xia Tang, Xiaojuan Song, Fang Yang, Xiaojuan Chen, Qianmeng Lin, Weibin Lin, Yang Zhou, ZhengChao Tu, Yongheng Chen, Zhang Zhang, Xiaoyun Lu
Abstract
MET alterations have been validated as a driven factor in NSCLC and gastric cancers. The c-Met inhibitors, capmatinib, tepotinib, and savolitinib, are only approved for the treatment of NSCLC harboring exon 14 skipping mutant MET. We used a molecular hybridization in conjunction with macrocyclization strategy for structural optimization to obtain a series of 2-(2-(quinolin-6-yl)ethyl)pyridazin-3(2H)-one derivatives as new c-Met inhibitors. One of the macrocyclic compounds, D6808, potently inhibited c-Met kinase and MET-amplified Hs746T gastric cancer cells with IC50 values of 2.9 and 0.7 nM, respectively. It also strongly suppressed Ba/F3-Tpr-Met cells harboring resistance-relevant mutations (F1200L/M1250T/H1094Y/F1200I/L1195V) with IC50 values of 4.2, 3.2, 1.0, 39.0, and 33.4 nM, respectively. Furthermore, D6808 exhibited extraordinary target specificity in a Kinome profiling against 373 wild-type kinases and served as a promising macrocycle-based compound for further anticancer drug development.
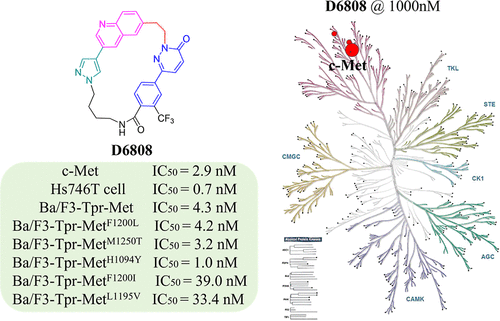
Overview
From C. Wang, Y. Chen, Z. Zhang, X. Lu et al. A macrocyclic c-Met inhibitor is more potent and selective that the acyclic version, is biologically active, but suffers from poor PK, … almost there.

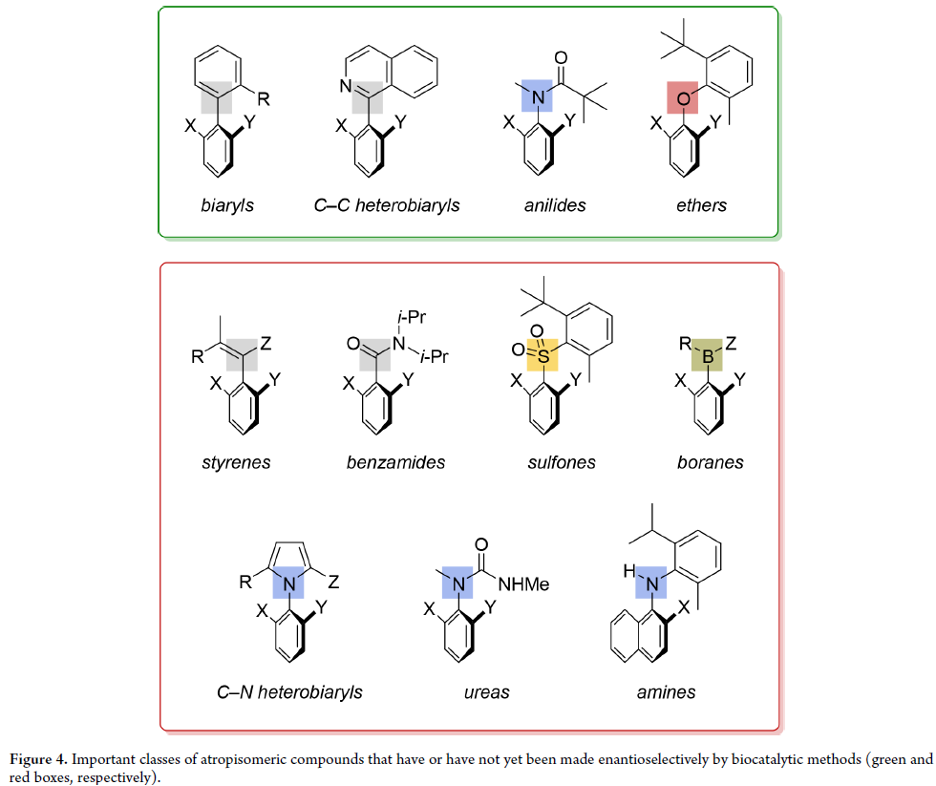
Biocatalytic Enantioselective Synthesis of Atropisomers
Authors: Olivia F. B. Watts, Jordan Berreur, Beatrice S. L. Collins, Jonathan Clayden
Abstract
Conspectus
Atropisomeric compounds are found extensively as natural products, as ligands for asymmetric transition-metal catalysis, and increasingly as bioactive and pharmaceutically relevant targets. Their enantioselective synthesis is therefore an important ongoing research target. While a vast majority of known atropisomeric structures are (hetero)biaryls, which display hindered rotation around a C–C single bond, our group’s long-standing interest in the control of molecular conformation has led to the identification and stereoselective preparation of a variety of other classes of “nonbiaryl” atropisomeric compounds displaying restricted rotation around C–C, C–N, C–O, and C–S single bonds.
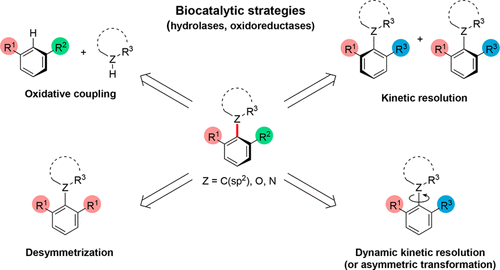
Overview
From O. F. B. Watts, J. Clayden et al. Another asymmetric synthesis review for aryl-atropisomers. This one has a list of potential compounds in the family yet to be figured out.