This blog series presents links to new literature on topics of Medicinal and Organic Chemistry.
It is derived from surveys of the Table of Contents for the Journal of Medicinal Chemistry, ACS Medicinal Chemistry Letters, Journal of Organic Chemistry, Journal of the American Chemical Society, Organic Letters, Organic Process Research and Development, Bioconjugate Chemistry, ACS Chemical Biology, Accounts of Chemical Research, Chemical Reviews ,and others with overviews provided by the blog author.

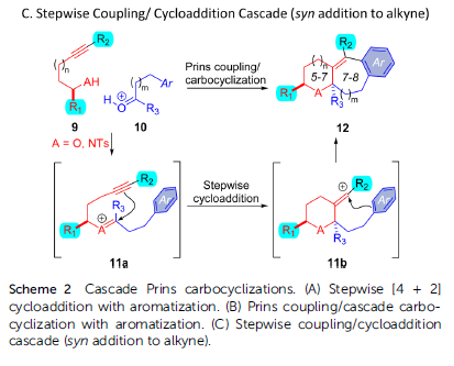
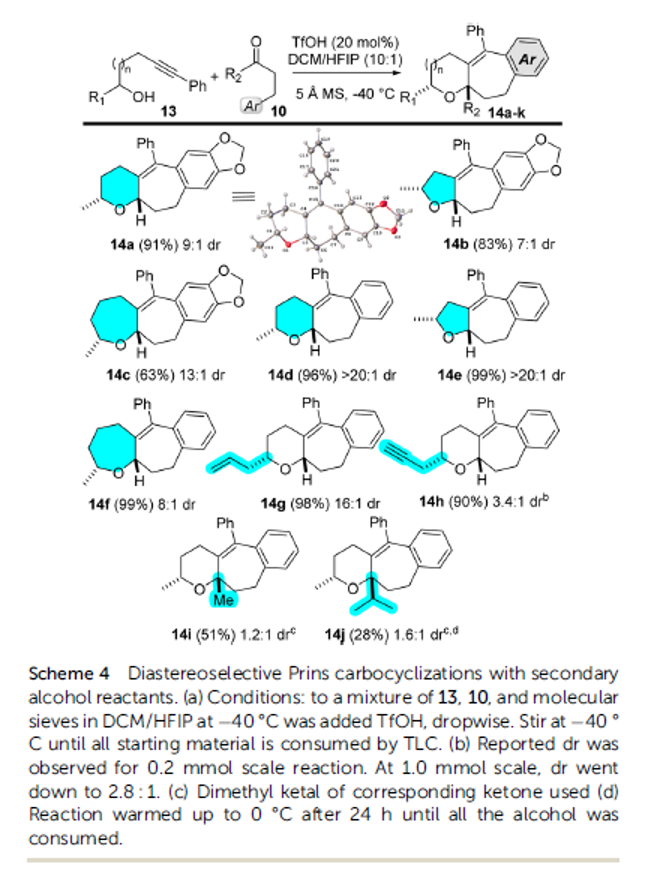
Alkynyl Prins carbocyclization cascades for the synthesis of linear-fused heterocyclic ring systems
Authors: Jackson J. Hernandez and Alison J. Frontier
Abstract
We report a Brønsted acid-catalyzed carbocyclization cascade, featuring condensation of an alcohol/sulfonamide with an aldehyde followed by an intramolecular three-component coupling involving an alkyne, an oxocarbenium/iminium ion, and an arene. A formal cycloaddition is embedded in the cationic cascade, which enables the synthesis of a wide range of fused heterotricycles. The diastereoselectivity of the cascade is studied using secondary alcohols/sulfonamides with different carbonyl partners. The described method results in the preparation of synthetically versatile scaffolds with ample opportunity for further derivatization at the resulting tetrasubstituted olefin, or by inclusion of other functionalizable motifs from the starting materials. It is worth noting that this chemistry also facilitates the synthesis of piperidines and 1,4-oxazepanes, as well as the inclusion of indoles and benzofurans, which are privileged motifs for medicinal chemistry. Herein we present the generality of this approach and some chemical transformations that can be achieved with our substrates.
Overview
From J. J. Hernandez and A. J. Frontier. This presents an intermolecular alkynyl Prins reaction that forms 3 bonds and at least 3 fused rings, 1 aryl and two aliphatic. It seems a promising approach to defined cyclic architectures that may be manipulated to have acceptable physical properties for medicinal chemistry. Here is one example table.
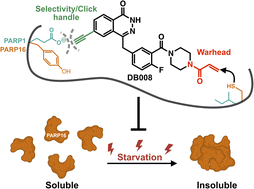
Structure-guided design and characterization of a clickable, covalent PARP16 inhibitor
Authors: Daniel S. Bejan, Sunil Sundalam, Haihong Jin, Rory K. Morgan, Ilsa T. Kirby,Ivan R. Siordia, Barr Tivon, Nir London and Michael S. Cohen
Abstract
PARP16—the sole ER-resident PARP family member—is gaining attention as a potential therapeutic target for cancer treatment. Nevertheless, the precise function of the catalytic activity of PARP16 is poorly understood. This is primarily due to the lack of inhibitors that are selective for PARP16 over other PARP family members. Herein, we describe a structure-guided strategy for generating a selective PARP16 inhibitor by incorporating two selectivity determinants into a phthalazinone pan-PARP inhibitor scaffold: (i) an acrylamide-based inhibitor (DB008) designed to covalently react with a non-conserved cysteine (Cys169, human numbering) in the NAD+ binding pocket of PARP16 and (ii) a dual-purpose ethynyl group designed to bind in a unique hydrophobic cavity adjacent to the NAD+ binding pocket as well as serve as a click handle. DB008 exhibits good selectivity for PARP16 versus other PARP family members. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) confirmed that covalent labeling of PARP16 by DB008 in cells is dependent on Cys169. DB008 exhibits excellent proteome-wide selectivity at concentrations required to achieve saturable labeling of endogenous PARP16. In-cell competition labeling experiments using DB008 provided a facile strategy for evaluating putative PARP16 inhibitors. Lastly, we found that PARP16 is sequestered into a detergent-insoluble fraction under prolonged amino acid starvation, and surprisingly, treatment with PARP16 inhibitors prevented this effect. These results suggest that the catalytic activity of PARP16 regulates its solubility in response to nutrient stress.
Overview
From D. S. Bejan, M. S. Cohen et al.. This discloses the first covalent inhibitor for a PARP family enzyme, PARP16. PARP16 has a unique CYS169 that the group was able to design an acrylamide group to form a covalent bond with. The compound was made further selective by adding a hydrophobic group, an alkyne which is less tolerated by other PARPs that have hydrophilic residues where PARP16 is hydrophobic. The alkyne also served the purpose of proof of the covalent connection after the complex was click-reacted with TAMRA-azide.

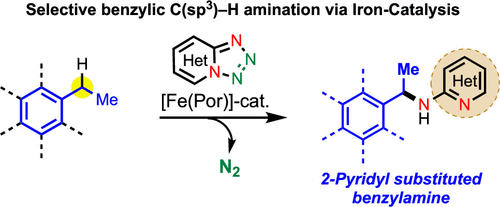
Iron-Catalyzed Intermolecular Amination of Benzylic C(sp3)–H Bonds
Authors: Hillol Khatua, Subrata Das, Sima Patra, Sandip Kumar Das, Satyajit Roy, and Buddhadeb Chattopadhyay
Abstract
A catalytic system for intermolecular benzylic C(sp3)–H amination is developed utilizing 1,2,3,4-tetrazole as a nitrene precursor via iron catalysis. This method enables direct installation of 2-aminopyridine into the benzylic and heterobenzylic position. The method selectively aminates 2° benzylic C(sp3)–H bond over the 3° and 1° benzylic C(sp3)–H bonds. Experimental studies reveal that the C(sp3)–H amination undergoes via the formation of a benzylic radical intermediate. This study reports the discovery of new method for 2-pyridine substituted benzylamine synthesis using inexpensive, biocompatible base metal catalysis that should have wide application in the context of medicinal chemistry and drug discovery.
Overview
From H. Khatua, B. Chattopadhyay et al. Using tetrazolo[1,5-a]pyridine as a 2-nitrenylpyridine precursor, a benzylic CH insertion reaction was developed that relies on a porphyrin catalyst. It is a little nitchy since it is limited to addition of 2-aminopyridine to benzylic positions. Who knows, it could lead to other more diverse benzylic amination in the future. Here is one table of results.
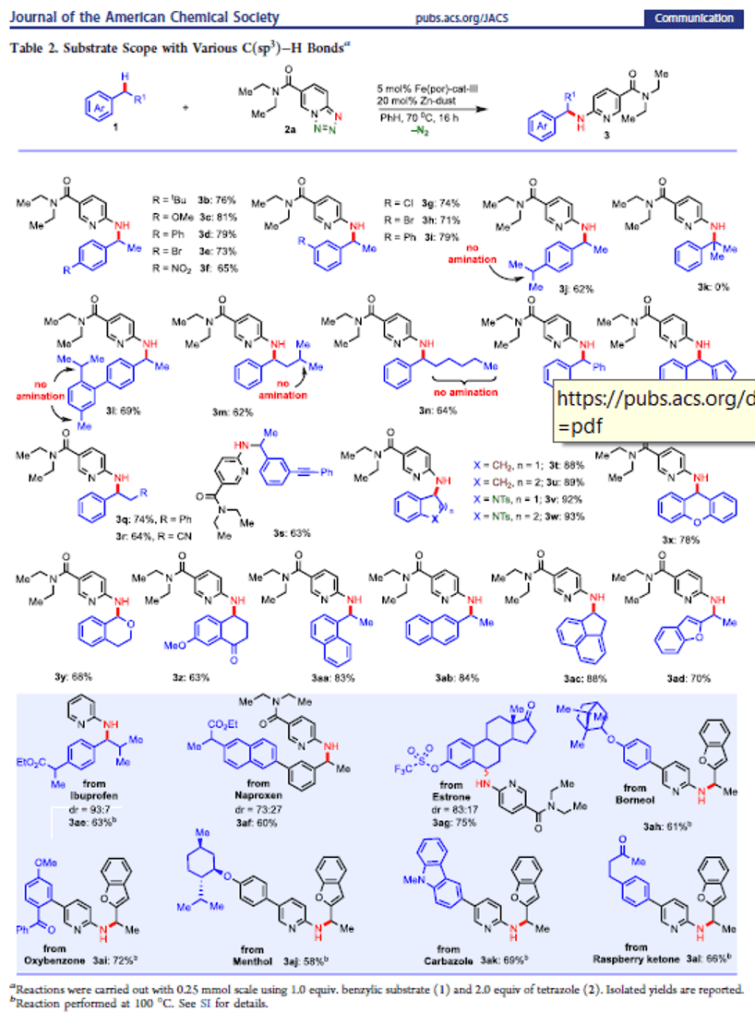

Synthesis and Characterization of NUC-7738, an Aryloxy Phosphoramidate of 3′-Deoxyadenosine, as a Potential Anticancer Agent
Authors: Michaela Serpi, Valentina Ferrari, Christopher McGuigan, Essam Ghazaly, Chris Pepper
Abstract
3′-Deoxyadenosine (3′-dA, Cordycepin, 1) is a nucleoside analogue with anticancer properties, but its clinical development has been hampered due to its deactivation by adenosine deaminase (ADA) and poor cellular uptake due to low expression of the human equilibrative transporter (hENT1). Here, we describe the synthesis and characterization of NUC-7738 (7a), a 5′-aryloxy phosphoramidate prodrug of 3′-dA. We show in vitro evidence that 7a is an effective anticancer drug in a panel of solid and hematological cancer cell lines, showing its preferential cytotoxic effects on leukemic stem cells. We found that unlike 3′-dA, the activity of 7a was independent of hENT1 and kinase activity. Furthermore, it was resistant to ADA metabolic deactivation. Consistent with these findings, 7a showed increased levels of intracellular 3′-deoxyadenosine triphosphate (3′-dATP), the active metabolite. Mechanistically, levels of intracellular 3′-dATP were strongly associated with in vitro potency. NUC-7738 is now in Phase II, dose-escalation study in patients with advanced solid tumors.
Overview
From M. Serpi et al. A study of ProTide 5’-monophosphate prodrugs of 3’-deoxyadenosine monophosphate proves they can be an effective tool for delivery of this anticancer drug to tumor sites. This molecule is in PII clinical studies for this purpose.

Diphenylsilylsilanolates Enable the Transfer of a Wide Range of Silyl Groups
Authors: Hiroki Yamagishi, Fuyuki Harata, Jun Shimokawa, Hideki Yorimitsu
Abstract
Development of silylating reagents that can transfer a wide range of silyl groups has been a long-standing challenge. Herein we report sodium diphenylsilylsilanolates as new stable and handy silylating reagents that could be synthesized from chlorosilanes. The new reagents retain the ability of dimethylsilylsilanolates for the delivery of a variety of silyl groups in palladium-catalyzed silylation of aryl bromides irrespective of the steric and electronic properties of silyl groups to be transferred.
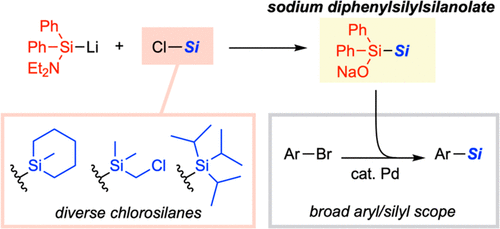
Overview
From H. Yamagishi, F. Harata, J. Shimokawa, H. Yorimitsu. This is a Pd-cross-coupling method to connect diverse Si- and Ge-groups to aryl bromides. Not only is there the isostere element but the Ge reagents could prove useful for further chemistry. Here is the example table.



